
Identification and Functional Analysis of Transcriptome Profiles, Long Non-Coding RNAs, Single-Nucleotide Polymorphisms, and Alternative Splicing from the Oocyte to the Preimplantation Stage of Sheep by Single-Cell RNA Sequencing
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Sample Collection
2.2. RNA Isolation, Library Preparation, and Sequencing
2.3. Quality Control and Assembly of Transcriptome Data
2.4. RNA-Seq Reads Mapping
2.5. Estimation of Transcript Abundance and Differential Expression and Principal Component Analysis (PCA)
2.6. WGCNA Analysis
2.7. Transcript Assembly and Identification of Candidate lncRNAs
2.8. Classification of lncRNAs
2.9. Quantification and Differential Expression Analysis
2.10. Predictions of Cis and Trans-Target Genes
2.11. SNP Analysis
2.12. AS Data Collection
2.13. GO and KEGG Enrichment Analysis
2.14. Statistical Analysis
3. Results
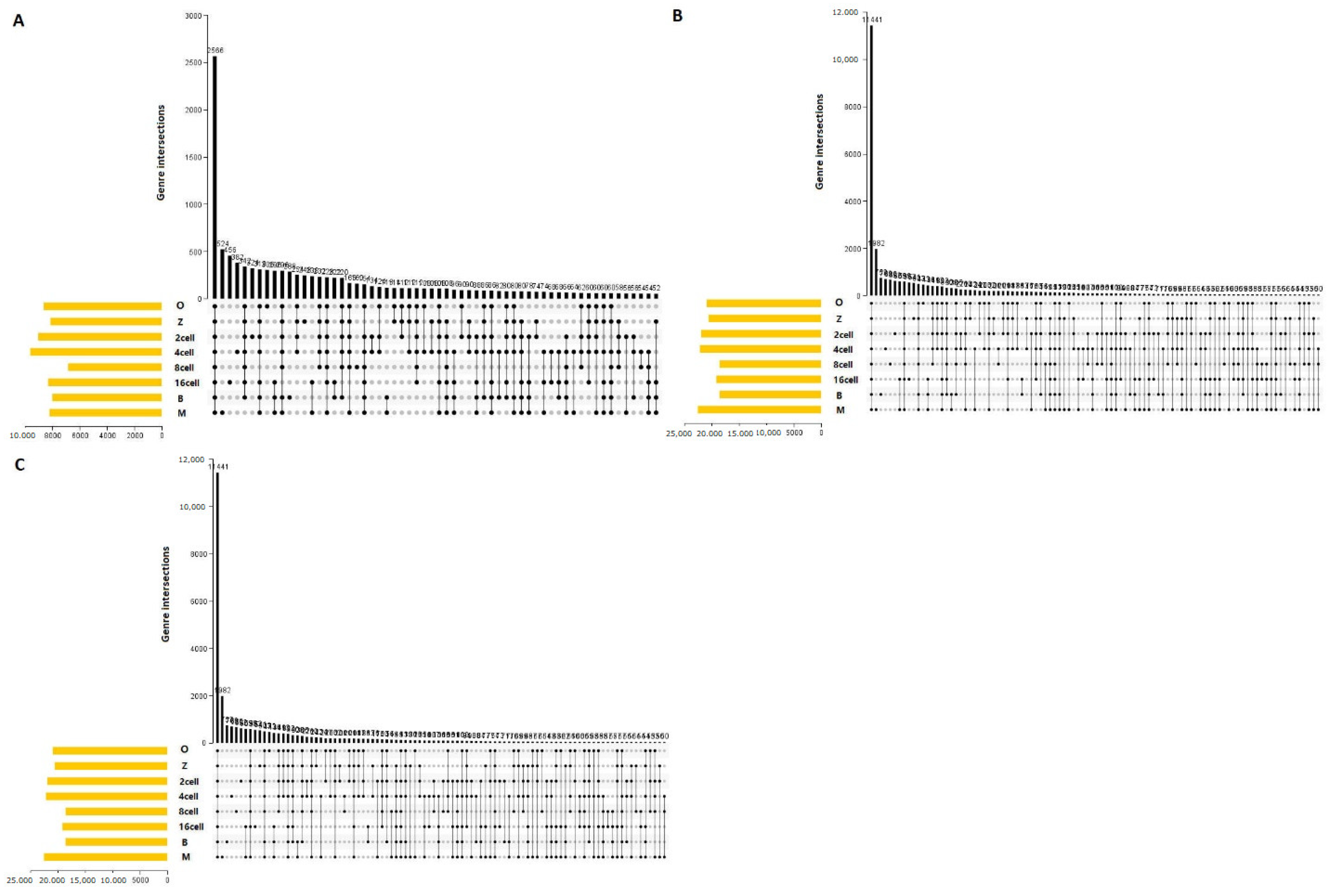
3.1. Transcriptome Profiles
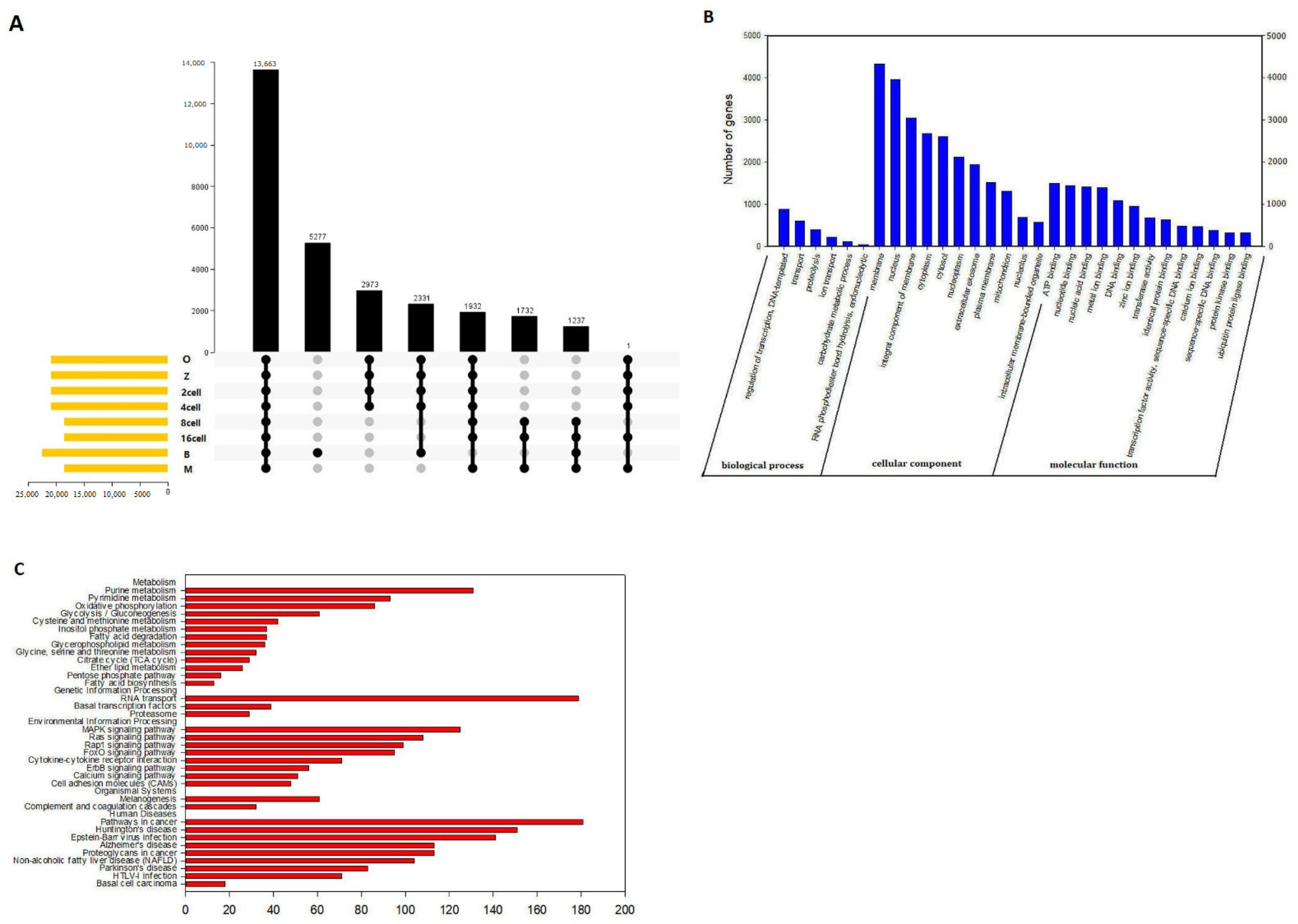
3.2. Differentially Expressed Genes
3.3. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes Analyses of DEGs
3.4. Genome-Wide Discovery and Identification of lncRNAs
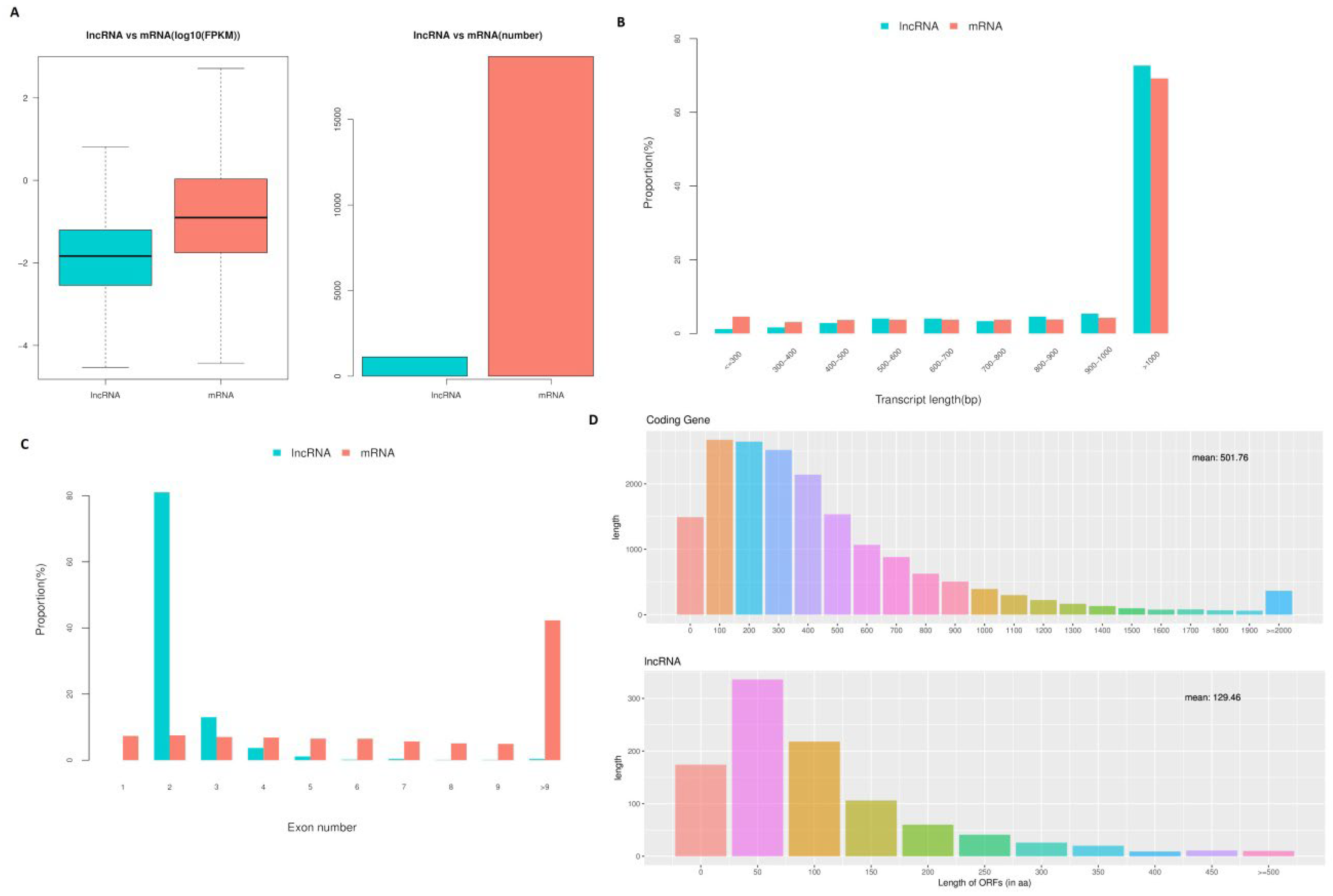
3.5. Features of lncRNAs
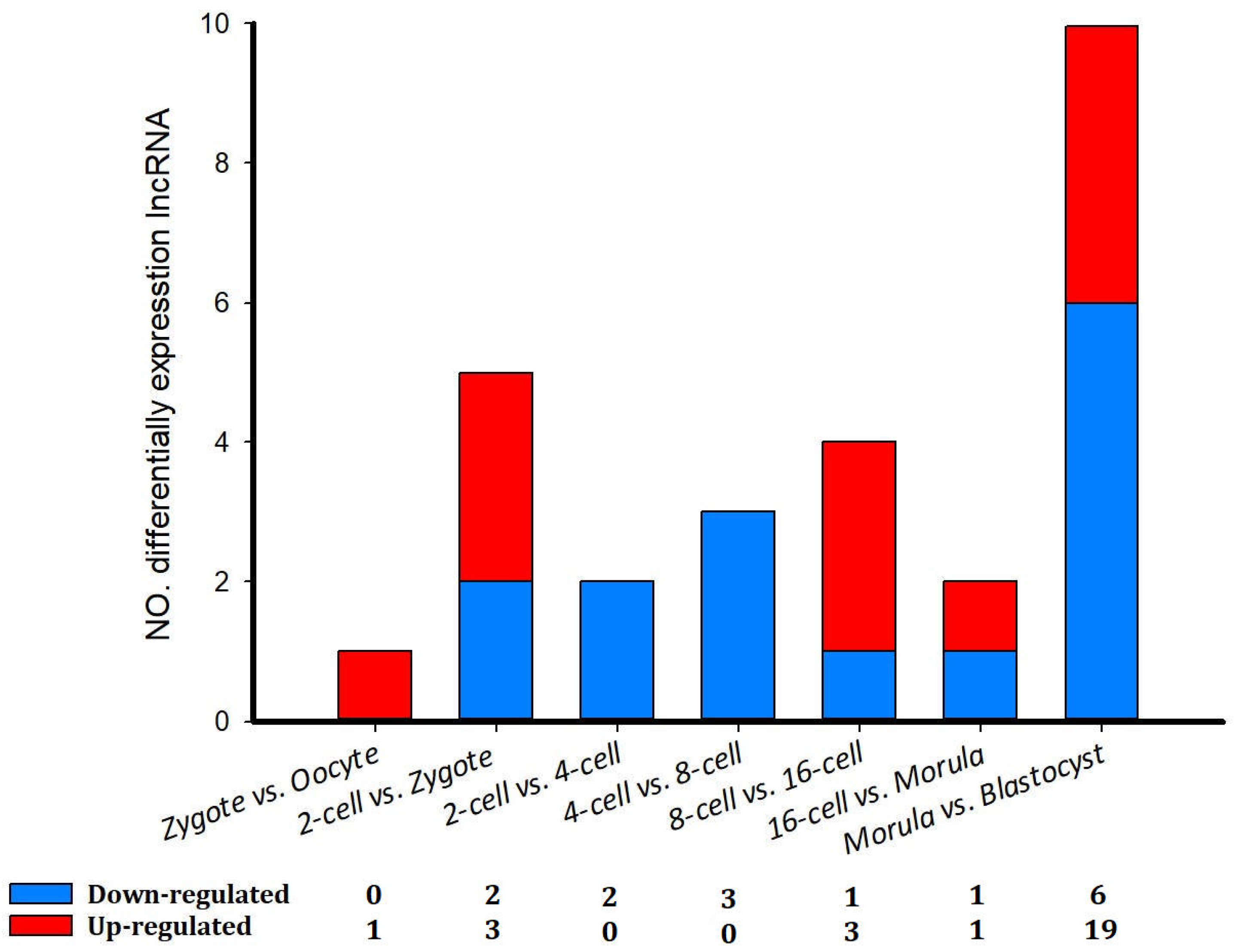
3.6. Differentially Expressed lncRNAs
3.7. Cis- and Trans-Target Genes of lncRNAs
3.8. Functional Analysis of Differentially Expressed lncRNAs
3.9. Distribution of Different SNP and Indel Types in Sheep from Oocyte to Blastocyst Development
3.10. SNP and Indel Functional Annotation
3.11. Overview of AS Events in Sheep from Oocyte to Blastocyst Development
3.12. Associated AS Events
3.13. Molecular Characteristics of the Most Important AS Events
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, K.; Huang, K.; Luo, Y.; Li, S. Identification and functional analysis of long non-coding RNAs in mouse cleavage stageembryonic development based on single cell transcriptome data. BMC Genom. 2014, 15, 845. [Google Scholar] [CrossRef]
- Daughtry, B.L.; Rosenkrantz, J.L.; Lazar, N.H.; Fei, S.S.; Redmayne, N.; Torkenczy, K.A.; Adey, A.; Yan, M.; Gao, L.; Park, B.; et al. Single-cell sequencing of primate preimplantation embryos reveals chromosome elimination via cellular fragmentation and blastomere exclusion. Genome Res. 2019, 29, 367–382. [Google Scholar] [CrossRef]
- Cheung, V.G.; Spielman, R.S. The genetics of variation in gene expression. Nat. Genet. 2002, 32, S522–S525. [Google Scholar] [CrossRef]
- Cheung, V.G.; Bruzel, A.; Burdick, J.T.; Morley, M.; Devlin, J.L.; Spielman, R.S. Monozygotic twins reveal germline contribution to allelic expression differences. Am. J. Hum. Genet. 2008, 82, 1357–1360. [Google Scholar] [CrossRef]
- Zhang, X.; Li, T.; Zhang, L.; Jiang, L.; Cui, T.; Yuan, X.; Wang, C.; Liu, Z.; Zhang, Y.; Li, W.; et al. Individual blastomeres of 4- and 8-cell embryos have ability to develop into a full organism in mouse. J. Genet. Genom. 2018, 45, 677–680. [Google Scholar] [CrossRef]
- Daigneault, B.W.; Rajput, S.; Smith, G.W.; Ross, P.J. Required for Expanded Bovine Blastocyst Formation. Sci. Rep. 2018, 8, 7753. [Google Scholar] [CrossRef]
- Deng, M.; Liu, Z.; Ren, C.; Zhang, G.; Pang, J.; Zhang, Y.; Wang, F.; Wan, Y. Long noncoding RNAs exchange during zygotic genome activation in goat. Biol. Reprod. 2018, 99, 707–717. [Google Scholar] [CrossRef]
- Yan, L.; Yang, M.; Guo, H.; Yang, L.; Wu, J.; Li, R.; Liu, P.; Lian, Y.; Zheng, X.; Yan, J.; et al. Single-cell RNA-Seq. profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol. 2013, 20, 1131–1139. [Google Scholar] [CrossRef]
- Blakeley, P.; Fogarty, N.M.; del Valle, I.; Wamaitha, S.E.; Hu, T.X.; Elder, K.; Snell, P.; Christie, L.; Robson, P.; Niakan, K.K. Defining the three cell lineages of the human blastocyst by single-cell RNA-seq. Development 2015, 142, 3151–3165. [Google Scholar] [CrossRef]
- Tang, F.; Lao, K.; Surani, M.A. Development and applications of single-cell transcriptome analysis. Nat. Methods 2011, 8, S6–S11. [Google Scholar] [CrossRef]
- Liu, X.M.; Wang, Y.K.; Liu, Y.H.; Yu, X.X.; Wang, P.C.; Li, X.; Du, Z.Q.; Yang, C.X. Single-cell transcriptome sequencing reveals that cell division cycle 5-like protein is essential for porcine oocyte maturation. J. Biol. Chem. 2018, 293, 1767–1780. [Google Scholar] [CrossRef]
- Yin, X.Y.; Cheng, G.H.; Guo, H.Y.; Wang, Q.; Li, Y.J.; Zhang, H. Single cell transcriptome profiling revealed differences in gene expression during oocyte maturation in Haimen white goats. Genet. Mol. Res. 2017, 16, 1–12. [Google Scholar] [CrossRef]
- Svoboda, P. Long and small noncoding RNAs during oocyte-to-embryo transition in mammals. Biochem. Soc. Trans. 2017, 45, 1117–1124. [Google Scholar] [CrossRef]
- Abbastabar, M.; Sarfi, M.; Golestani, A.; Khalili, E. lncRNA involvement in hepatocellular carcinoma metastasis ands prognosis. EXCLI J. 2018, 17, 900–913. [Google Scholar]
- Youness, R.A.; Gad, M.Z. Long non-coding RNAs: Functional regulatory players in breast cancer. Noncoding RNA Res. 2019, 4, 36–44. [Google Scholar] [CrossRef]
- Karlic, R.; Ganesh, S.; Franke, V.; Svobodova, E.; Urbanova, J.; Suzuki, Y.; Aoki, F.; Vlahovicek, K.; Svoboda, P. Long non-coding RNA exchange during the oocyte-to-embryo transition in mice. DNA Res. 2017, 2, 129–141. [Google Scholar]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef]
- Rivera, R.M.; Ross, J.W. Epigenetics in fertilization and preimplantation embryo development. Prog. Biophys. Mol. Biol. 2013, 113, 423–432. [Google Scholar] [CrossRef]
- Volders, P.J.; Verheggen, K.; Menschaert, G.; Vandepoele, K.; Martens, L.; Vandesompele, J.; Mestdagh, P. An update on LNCipedia: A database for annotated human lncRNA sequences. Nucleic Acids Res. 2015, 43, 4363–4364. [Google Scholar] [CrossRef]
- Veselovska, L.; Smallwood, S.A.; Saadeh, H.; Stewart, K.R.; Krueger, F.; Maupetit-Méhouas, S.; Arnaud, P.; Tomizawa, S.; Andrews, S.; Kelsey, G. Deep sequencing and de novo assembly of the mouse oocyte transcriptome define the contribution of transcription to the DNA methylation landscape. Genome Biol. 2015, 16, 209. [Google Scholar] [CrossRef]
- Wu, F.; Liu, Y.; Wu, Q.; Li, D.; Zhang, L.; Wu, X.; Wang, R.; Zhang, D.; Gao, S.; Li, W. Long non-coding RNAs potentially function synergistically in the cellular reprogramming of SCNT embryos. BMC Genom. 2018, 19, 631. [Google Scholar] [CrossRef]
- Caballero, J.; Gilbert, I.; Fournier, E.; Gagné, D.; Scantland, S.; Macaulay, A.; Robert, C. Exploring the function of long non-coding RNA in the development of bovine early embryos. Reprod. Fertil. Dev. 2014, 27, 40–52. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 11–12. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Min, B.; Park, J.S.; Kang, Y.K. Determination of Oocyte-Manipulation, Zygote-Manipulation, and Genome-Reprogramming Effects on the Transcriptomes of Bovine Blastocysts. Front. Genet. 2018, 9, 143. [Google Scholar] [CrossRef]
- Xie, D.; Chen, C.C.; Ptaszek, L.M.; Xiao, S.; Cao, X.; Fang, F.; Ng, H.H.; Lewin, H.A.; Cowan, C.; Zhong, S. Rewirable gene regulatory networks in the preimplantation embryonic development of three mammalian species. Genome Res. 2010, 20, 804–815. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, N.; Cai, R.; Zhao, F.; Xiong, Y.; Li, X.; Wang, A.; Lin, P.; Jin, Y. Genome-wide analysis and functional prediction of long non-coding RNAs in mouse uterus during the implantation window. Oncotarget 2017, 8, 84360–84372. [Google Scholar] [CrossRef]
- Al-Tobasei, R.; Ali, A.; Leeds, T.D.; Liu, S.; Palti, Y.; Kenney, B.; Salem, M. Identification of SNPs associated with muscle yield and quality traits using allelic-imbalanceanalyses of pooled RNA-Seq samples in rainbow trout. BMC Genom. 2017, 18, 582. [Google Scholar] [CrossRef]
- Neininger, K.; Marschall, T.; Helms, V. SNP and indel frequencies at transcription start sites and at canonical and alternative translation initiation sites in the human genome. PLoS ONE 2019, 14, e0214816. [Google Scholar] [CrossRef]
- Raineri, E.; Ferretti, L.; Esteve-Codina, A.; Nevado, B.; Heath, S.; Pérez-Enciso, M. SNP calling by sequencing pooled samples. BMC Bioinform. 2012, 13, 239. [Google Scholar] [CrossRef]
- Piskol, R.; Ramaswami, G.; Li, J.B. Reliable identification of genomic variants from RNA-seq data. Am. J. Hum. Genet. 2013, 93, 641–651. [Google Scholar] [CrossRef]
- Berthelot, C.; Brunet, F.; Chalopin, D.; Juanchich, A.; Bernard, M.; Noel, B.; Bento, P.; Da Silva, C.; Labadie, K.; Alberti, A.; et al. The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat. Commun. 2014, 5, 3657. [Google Scholar] [CrossRef]
- Petropoulos, S.; Edsgärd, D.; Reinius, B.; Deng, Q.; Panula, S.P.; Codeluppi, S.; Plaza Reyes, A.; Linnarsson, S.; Sandberg, R.; Lanner, F. Single-Cell RNA-Seq Reveals Lineage and X Chromosome Dynamics in Human Preimplantation Embryos. Cell 2016, 165, 1012–1026. [Google Scholar] [CrossRef]
- Macaulay, I.C.; Voet, T. Single cell genomics: Advances and future perspectives. PLoS Genet. 2014, 10, e1004126. [Google Scholar] [CrossRef]
- Liu, Q.; Zhou, J.-P.; Li, B.; Huang, Z.-C.; Dong, H.-Y.; Li, G.-Y.; Zhou, K.; Nie, S.-L. Basic transcription factor 3 is involved in gastric cancer development and progression. World J. Gastroenterol. 2013, 19, 4495–4503. [Google Scholar] [CrossRef]
- Hosseini, S.; Hosseini, S.; Salehi, M. Upregulation of Toll-like receptor 4 through anti-miR-Let-7a enhances blastocyst attachment to endometrial cells in mice. J. Cell. Physiol. 2020, 235, 9752–9762. [Google Scholar] [CrossRef]
- Lavagi, I.; Krebs, S.; Simmet, K.; Beck, A.; Zakhartchenko, V.; Wolf, E.; Blum, H. Single-cell RNA sequencing reveals developmental heterogeneity of blastomeres during major genome activation in bovine embryos. Sci. Rep. 2018, 8, 4071. [Google Scholar] [CrossRef]
- Razza, E.M.; Sudano, M.J.; Fontes, P.K.; Franchi, F.F.; Belaz, K.R.A.; Santos, P.H.; Castilho, A.C.S.; Rocha, D.F.O.; Eberlin, M.N.; Machado, M.F.; et al. Treatment with cyclic adenosine monophosphate modulators prior to in vitro maturation alters the lipid composition and transcript profile of bovine cumulus-oocyte complexes and blastocysts. Reprod. Fertil. Dev. 2018, 30, 1314–1328. [Google Scholar] [CrossRef]
- Zhang, F.; Lupski, J.R. Non-coding genetic variants in human disease. Hum. Mol. Genet. 2015, 24, R102–R110. [Google Scholar] [CrossRef]
- Do, D.V.; Strauss, B.; Cukuroglu, E.; Macaulay, I.; Wee, K.B.; Hu, T.X.; Igor, R.L.M.; Lee, C.; Harrison, A.; Butler, R.; et al. SRSF3 maintains transcriptome integrity in oocytes by regulation of alternative splicing and transposable elements. Cell Discov. 2018, 4, 33. [Google Scholar] [CrossRef]
- Braunschweig, U.; Gueroussov, S.; Plocik, A.M.; Graveley, B.R.; Blencowe, B.J. Dynamic integration of splicing within gene regulatory pathways. Cell 2013, 152, 1252–1269. [Google Scholar] [CrossRef]
- Daguenet, E.; Dujardin, G.; Valcarcel, J. The pathogenicity of splicing defects: Mechanistic insights into pre-mRNA processing inform novel therapeutic approaches. EMBO Rep. 2015, 16, 1640–1655. [Google Scholar] [CrossRef]
- Hayashi, Y.; Mori, M.; Igarashi, K.; Tanaka, K.; Takehara, A.; Ito-Matsuoka, Y.; Kanai, A.; Yaegashi, N.; Soga, T.; Matsui, Y. Proteomic and metabolomic analyses uncover sex-specific regulatory pathways in mouse fetal germline differentiation. Biol. Reprod. 2020, 103, 717–735. [Google Scholar] [CrossRef]
- Revil, T.; Gaffney, D.; Dias, C.; Majewski, J.; Jerome-Majewska, L.A. Alternative splicing is frequent during early embryonic development in mouse. BMC Genom. 2010, 11, 399. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, R.; Wei, B.; Peng, C.; Wang, L.; Hu, G.; Kong, D.; Du, C. Candidate Biomarkers and Molecular Mechanism Investigation for Glioblastoma MultiformeUtilizing WGCNA. Biomed. Res. Int. 2018, 2018, 4246703. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | No. of Sample | No. of Single Cells |
|---|---|---|
| Oocyte | 3 | 3 |
| Zygote | 3 | 3 |
| 2-cell | 3 | 3 |
| 4-cell | 3 | 3 |
| 8-cell | 3 | 3 |
| 16-cell | 3 | 3 |
| Morula | 3 | 3 |
| Blastocyst | 3 | 3 |
| Total | 24 | 24 |
| Stage | No. of Genes (FPKM > 0.1) |
|---|---|
| Oocyte | 24,729 |
| Zygote | 25,455 |
| 2-cell | 26,714 |
| 4-cell | 28,948 |
| 8-cell | 25,920 |
| 16-cell | 24,341 |
| Morula | 22,242 |
| Blastocyst | 31,157 |
| Sample | Raw Reads | Clean Reads | Clean Bases | Error Rate (%) | GC Content (%) |
|---|---|---|---|---|---|
| oocyte | 102,383,290 | 86,741,418 | 13.01 G | 0.04 | 43.50 |
| Zygote | 111,921,426 | 87,081,144 | 13.06 G | 0.02 | 44.50 |
| 2-cell | 123,273,652 | 95,679,634 | 14.35 G | 0.04 | 44 |
| 4-cell | 108,522,186 | 96,419,102 | 14.46 G | 0.03 | 44 |
| 8-cell | 142,178,172 | 123,561,104 | 18.53 G | 0.02 | 45 |
| 16-cell | 133,023,048 | 90,477,914 | 13.57 G | 0.04 | 43.5 |
| morula | 122,556,646 | 90,506,796 | 13.58 G | 0.02 | 44.5 |
| blastula | 141,488,370 | 105,354,406 | 15.80 G | 0.03 | 44 |
| Functional Class | SNP | INDEL | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Oocyte | Zygote | 2-Cell | 4-Cell | 8-Cell | 16-Cell | Morula | Blastocyst | Oocyte | Zygote | 2-Cell | 4-Cell | 8-Cell | 16-Cell | Morula | Blastocyst | |
| Intergenic | 79,516 | 70,858 | 99,129 | 115,926 | 72,615 | 78,782 | 66,420 | 224,557 | 5650 | 5260 | 7088 | 8129 | 6163 | 4939 | 4765 | 15,130 |
| Intronic | 47,852 | 40,178 | 60,970 | 67,126 | 37,762 | 64,925 | 39,054 | 201,642 | 3104 | 2831 | 3973 | 4172 | 2793 | 3580 | 2566 | 15,373 |
| exonic | 28,898 | 24,940 | 33,951 | 35,201 | 26,715 | 32,414 | 27,569 | 66,325 | 336 | 298 | 438 | 411 | 415 | 326 | 440 | 512 |
| 3′UTR | 3991 | 4763 | 5879 | 5761 | 4773 | 4822 | 3991 | 8356 | 502 | 564 | 666 | 651 | 598 | 547 | 577 | 862 |
| 5′UTR | 1075 | 904 | 1142 | 1269 | 964 | 1265 | 1075 | 2145 | 34 | 32 | 35 | 42 | 35 | 41 | 44 | 60 |
| upstream | 4343 | 3612 | 5330 | 5836 | 3937 | 5551 | 4343 | 9568 | 243 | 233 | 359 | 408 | 285 | 306 | 299 | 588 |
| downstream | 14,572 | 16,348 | 19,799 | 20,220 | 16,904 | 16,251 | 14,572 | 27,967 | 1845 | 1814 | 2268 | 2209 | 2072 | 1749 | 2010 | 2886 |
| splicing | 632 | 375 | 421 | 494 | 390 | 390 | 632 | 525 | 47 | 43 | 53 | 52 | 47 | 44 | 53 | 71 |
| Total number | 184,380 | 162,027 | 226,690 | 251,891 | 164,117 | 204,485 | 157,743 | 541,244 | 11,763 | 11,076 | 14,882 | 16,076 | 12,408 | 11,534 | 10,755 | 35,487 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Shi, Q.; Zhu, X.; Jin, L.; Lang, L.; Lyu, S.; Xin, X.; Huang, Y.; Yu, X.; Li, Z.; et al. Identification and Functional Analysis of Transcriptome Profiles, Long Non-Coding RNAs, Single-Nucleotide Polymorphisms, and Alternative Splicing from the Oocyte to the Preimplantation Stage of Sheep by Single-Cell RNA Sequencing. Genes 2023, 14, 1145. https://doi.org/10.3390/genes14061145
Zhang Z, Shi Q, Zhu X, Jin L, Lang L, Lyu S, Xin X, Huang Y, Yu X, Li Z, et al. Identification and Functional Analysis of Transcriptome Profiles, Long Non-Coding RNAs, Single-Nucleotide Polymorphisms, and Alternative Splicing from the Oocyte to the Preimplantation Stage of Sheep by Single-Cell RNA Sequencing. Genes. 2023; 14(6):1145. https://doi.org/10.3390/genes14061145
Chicago/Turabian StyleZhang, Zijing, Qiaoting Shi, Xiaoting Zhu, Lei Jin, Limin Lang, Shijie Lyu, Xiaoling Xin, Yongzhen Huang, Xiang Yu, Zhiming Li, and et al. 2023. "Identification and Functional Analysis of Transcriptome Profiles, Long Non-Coding RNAs, Single-Nucleotide Polymorphisms, and Alternative Splicing from the Oocyte to the Preimplantation Stage of Sheep by Single-Cell RNA Sequencing" Genes 14, no. 6: 1145. https://doi.org/10.3390/genes14061145