
Prognostic Values of Gene Copy Number Alterations in Prostate Cancer
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Cohort
2.2. Clinical and Genomic Data
2.3. Genomic Data Analyses
2.4. Study Approach and Principles
2.5. Statistical Analyses
3. Result
3.1. Clinicopathological Characteristics of PCa Cohort
3.2. Copy Number Segments and Genetic Markers
3.3. Gene Level Copy Number Alterations Are Associated with Disease Stage
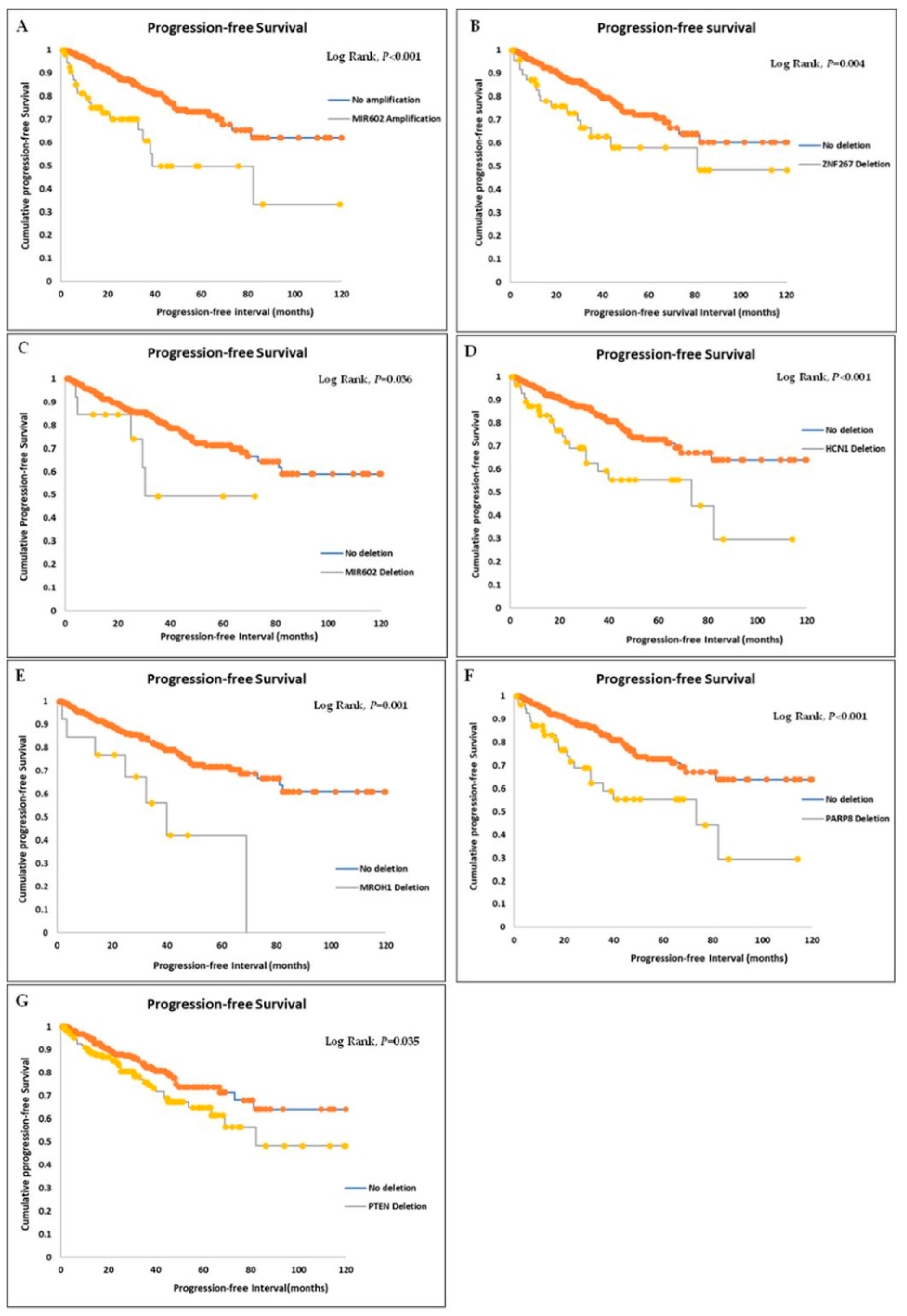
3.4. Survival Significance of Gene CNAs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Kulac, I.; Roudier, M.P.; Haffner, M.C. Molecular Pathology of Prostate Cancer. Surg. Pathol. Clin. 2021, 14, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Konishi, K.; Komatsu, T.; Ishiba, R. Quality of life after external beam radiotherapy for localized prostate cancer: Comparison with other modalities. Int. J. Urol. 2019, 26, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Bruner, D.W.; Pugh, S.L.; Lee, W.R.; Hall, W.A.; Dignam, J.J.; Low, D.; Swanson, G.P.; Shah, A.B.; Malone, S.; Michalski, J.M.; et al. Quality of life in patients with low-risk prostate cancer treated with hypofractionated vs conventional radiotherapy: A phase 3 randomized clinical trial. JAMA Oncol. 2019, 5, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M.; Chen, E.V.; Miller, R.C.; Greenberger, A.B. The Impact of Prostate Cancer Treatment on Quality of Life: A Narrative Review with a Focus on Randomized Data. Res. Rep. Urol. 2020, 12, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z. Distinguishing indolent from aggressive prostate cancer. Recent Results Cancer Res. 2014, 202, 141–147. [Google Scholar] [CrossRef]
- Hoogland, A.M.; Kweldam, C.F.; Van Leenders, G.J.L.H. Prognostic Histopathological and Molecular Markers on Prostate Cancer Needle-Biopsies: A Review. BioMed Res. Int. 2014, 2014, 341324. [Google Scholar] [CrossRef]
- Khoddami, M.; Khademi, Y.; Aghdam, M.K.; Soltanghoraee, H. Correlation between Gleason Scores in Needle Biopsy and Corresponding Radical Prostatectomy Specimens: A Twelve-Year Review. Iran. J. Pathol. 2016, 11, 120–126. [Google Scholar]
- Camacho, N.; Van Loo, P.; Edwards, S.; Kay, J.D.; Matthews, L.; Haase, K.; Clark, J.; Dennis, N.; Thomas, S.; Kremeyer, B.; et al. Appraising the relevance of DNA copy number loss and gain in prostate cancer using whole genome DNA sequence data. PLOS Genet. 2017, 13, e1007001. [Google Scholar] [CrossRef]
- Köseoğlu, H. Genetics in the Prostate Cancer. In Prostate Cancer; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef]
- Williams, J.L.; Greer, P.A.; Squire, J.A. Recurrent copy number alterations in prostate cancer: An in silico meta-analysis of publicly available genomic data. Cancer Genet. 2014, 207, 474–488. [Google Scholar] [CrossRef]
- Fraser, M.; Sabelnykova, V.Y.; Yamaguchi, T.N.; Heisler, L.E.; Livingstone, J.; Huang, V.; Shiah, Y.-J.; Yousif, F.; Lin, X.; Masella, A.P.; et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017, 541, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Grist, E.; Friedrich, S.; Brawley, C.; Mendes, L.; Parry, M.; Ali, A.; Haran, A.; Hoyle, A.; Gilson, C.; Lall, S.; et al. Accumulation of copy number alterations and clinical progression across advanced prostate cancer. Genome Med. 2022, 14, 102. [Google Scholar] [CrossRef]
- Hieronymus, H.; Murali, R.; Tin, A.; Yadav, K.; Abida, W.; Moller, H.; Berney, D.; Scher, H.; Carver, B.; Scardino, P.; et al. Tumor copy number alteration burden is a pan-cancer prognostic factor associated with recurrence and death. Elife 2018, 7, e37294. [Google Scholar] [CrossRef] [PubMed]
- Hieronymus, H.; Schultz, N.; Gopalan, A.; Carver, B.S.; Chang, M.T.; Xiao, Y.; Heguy, A.; Huberman, K.; Bernstein, M.; Assel, M.; et al. Copy number alteration burden predicts prostate cancer relapse. Proc. Natl. Acad. Sci. USA 2014, 111, 11139–11144. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Yoshimoto, M.; Cunha, I.W.; Coudry, R.A.; Fonseca, F.P.; Torres, C.H.; Soares, F.A.; Squire, J.A. FISH analysis of 107 prostate cancers shows that PTEN genomic deletion is associated with poor clinical outcome. Br. J. Cancer 2007, 97, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dai, B. PTEN genomic deletion defines favourable prognostic biomarkers in localized prostate cancer: A systematic review and meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 5430–5437. [Google Scholar]
- Krohn, A.; Diedler, T.; Burkhardt, L.; Mayer, P.S.; De Silva, C.; Meyer-Kornblum, M.; Kötschau, D.; Tennstedt, P.; Huang, J.; Gerhäuser, C.; et al. Genomic deletion of PTEN is associated with tumour progression and early PSA recurrence in erg fusion-positive and fusion-negative prostate cancer. Am. J. Pathol. 2012, 181, 401–412. [Google Scholar] [CrossRef]
- Troyer, D.A.; Jamaspishvili, T.; Wei, W.; Feng, Z.; Good, J.; Hawley, S.; Fazli, L.; McKenney, J.K.; Simko, J.; Hurtado-Coll, A.; et al. A multicenter study shows PTEN deletion is strongly associated with seminal vesicle involvement and extracapsular extension in localized prostate cancer. Prostate 2015, 75, 1206–1215. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef]
- Visakorpi, T.; Hyytinen, E.; Koivisto, P.; Tanner, M.; Keinänen, R.; Palmberg, C.; Palotie, A.; Tammela, T.; Isola, J.; Kallioniemi, O.P. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995, 9, 401–406. [Google Scholar] [CrossRef]
- Pearson, H.B.; Li, J.; Meniel, V.S.; Fennell, C.M.; Waring, P.; Montgomery, K.G.; Rebello, R.J.; Macpherson, A.A.; Koushyar, S.; Furic, L.; et al. Identification of Pik3ca Mutation as a Genetic Driver of Prostate Cancer That Cooperates with Pten Loss to Accelerate Progression and Castration-Resistant Growth. Cancer Discov. 2018, 8, 764–779. [Google Scholar] [CrossRef]
- Zafarana, G.; Ishkanian, A.S.; Malloff, C.A.; Locke, J.A.; Sykes, J.; Thoms, J.; Lam, W.L.; Squire, J.A.; Yoshimoto, M.; Ramnarine, V.R.; et al. Copy number alterations of c-MYC and PTEN are prognostic factors for relapse after prostate cancer radiotherapy. Cancer 2012, 118, 4053–4062. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Liu, W.; Xie, C.C.; Thomas, C.Y.; Kim, S.-T.; Lindberg, J.; Egevad, L.; Wang, Z.; Zhang, Z.; Sun, J.; Sun, J.; et al. Genetic markers associated with early cancer-specific mortality following prostatectomy. Cancer 2013, 119, 2405–2412. [Google Scholar] [CrossRef]
- Kindich, R.; Florl, A.R.; Kamradt, J.; Lehmann, J.; Müller, M.; Wullich, B.; Schulz, W.A. Relationship of NKX3.1 and MYC gene copy number ratio and DNA hypomethylation to prostate carcinoma stage. Eur. Urol. 2006, 49, 169–175. [Google Scholar] [CrossRef]
- Sirohi, D.; Devine, P.; Grenert, J.P.; Van Ziffle, J.; Simko, J.P.; Stohr, B.A. TP53 structural variants in metastatic prostatic carcinoma. PLoS ONE 2019, 14, e0218618. [Google Scholar] [CrossRef] [PubMed]
- Hamid, A.A.; Gray, K.P.; Shaw, G.; MacConaill, L.E.; Evan, C.; Bernard, B.; Loda, M.; Corcoran, N.M.; Van Allen, E.M.; Choudhury, A.D.; et al. Compound genomic alterations of TP53, PTEN, and RB1 tumor suppressors in localized and metastatic prostate cancer. Eur. Urol. 2019, 76, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Nientiedt, C.; Endris, V.; Jenzer, M.; Mansour, J.; Pour Sedehi, N.T.; Pecqueux, C.; Volckmar, A.-L.; Leichsenring, J.; Neumann, O.; Kirchner, M.; et al. High prevalence of DNA damage repair gene defects and TP53 alterations in men with treatment-naïve metastatic prostate cancer—Results from a prospective pilot study using a 37 gene panel. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 637.e17–637.e27. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.U.; Rider, L.; Nieto, C.; Romero, L.; Karimpour-Fard, A.; Loda, M.; Lucia, M.S.; Wu, M.; Shi, L.; Cimic, A.; et al. Coordinate loss of MAP3K7 and CHD1 promotes aggressive prostate cancer. Cancer Res. 2015, 75, 1021–1034. [Google Scholar] [CrossRef] [PubMed]
- Ross-Adams, H.; Lamb, A.D.; Dunning, M.J.; Halim, S.; Lindberg, J.; Massie, C.M.; Egevad, L.A.; Russell, R.; Ramos-Montoya, A.; Vowler, S.L.; et al. Integration of copy number and transcriptomics provides risk stratification in prostate cancer: A discovery and validation cohort study. EBioMedicine 2015, 2, 1133–1144. [Google Scholar] [CrossRef]
- Chung, J.H.; Dewal, N.; Sokol, E.; Mathew, P.; Whitehead, R.; Millis, S.Z.; Frampton, G.M.; Bratslavsky, G.; Pal, S.K.; Lee, R.J.; et al. Prospective Comprehensive Genomic Profiling of Primary and Metastatic Prostate Tumors. JCO Precis. Oncol. 2019, 3, 1–23. [Google Scholar] [CrossRef]
- Fraser, M.; Livingstone, J.; Wrana, J.L.; Finelli, A.; He, H.H.; van der Kwast, T.; Zlotta, A.R.; Bristow, R.G.; Boutros, P.C. Somatic driver mutation prevalence in 1844 prostate cancers identifies ZNRF3 loss as a predictor of metastatic relapse. Nat. Commun. 2021, 12, 6248. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, J.; Zhang, Y.; Yuan, M.; Yang, S.; Li, L.; Yang, H. Prognostic Values of CCNE1 Amplification and Overexpression in Cancer Patients: A Systematic Review and Meta-analysis. J. Cancer 2018, 9, 2397–2407. [Google Scholar] [CrossRef] [PubMed]
- Faisal, F.A.; Murali, S.; Kaur, H.; Vidotto, T.; Guedes, L.B.; Salles, D.C.; Kothari, V.; Tosoian, J.J.; Han, S.; Hovelson, D.H.; et al. CDKN1B deletions are associated with metastasis in African American men with clinically localized, surgically treated prostate cancer. Clin. Cancer Res. 2020, 26, 2595–2602. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.L.; Botta, G.; Gao, S.; Li, T.; Van Allen, E.M.; Treacy, D.J.; Cai, C.; He, H.H.; Sweeney, C.J.; Brown, M.; et al. PLZF, a tumor suppressor genetically lost in metastatic castration-resistant prostate cancer, is a mediator of resistance to androgen deprivation therapy. Cancer Res. 2015, 75, 1944–1948. [Google Scholar] [CrossRef]
- Irshad, S.; Bansal, M.; Castillo-Martin, M.; Zheng, T.; Aytes, A.; Wenske, S.; Le Magnen, C.; Guarnieri, P.; Sumazin, P.; Benson, M.C.; et al. A molecular signature predictive of indolent prostate cancer. Sci. Transl. Med. 2013, 5, 202ra122. [Google Scholar] [CrossRef]
- Ameri, A.; Alidoosti, A.; Hosseini, Y.; Parvin, M.; Emranpour, M.H.; Taslimi, F.; Salehi, E.; Fadavi, P. Prognostic value of promoter hypermethylation of retinoic acid receptor beta (RARB) and CDKN2A (p16/MTS1) in prostate cancer. Chin. J. Cancer Res. 2011, 23, 306–311. [Google Scholar] [CrossRef]
- Zhao, R.; Choi, B.Y.; Lee, M.-H.; Bode, A.M.; Dong, Z. Implications of Genetic and Epigenetic Alterations of CDKN2A (p16 INK4a) in Cancer. Ebiomedicine 2016, 8, 30–39. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Xu, L.; Zhang, J.; Cao, H. Analysis of the EGFR Amplification and CDKN2A Deletion Regulated Transcriptomic Signatures Reveals the Prognostic Significance of SPATS2L in Patients with Glioma. Front. Oncol. 2021, 11, 551160. [Google Scholar] [CrossRef] [PubMed]
- Catz, S.D.; Johnson, J.L. BCL2 in prostate cancer: A minireview. Apoptosis 2003, 8, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Renner, W.; Langsenlehner, U.; Krenn-Pilko, S.; Eder, P.; Langsenlehner, T. BCL2 genotypes and prostate cancer survival. Strahlenther. Onkol. 2017, 193, 466. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Qin, J.; Lee, H.-J.; Wu, S.-P.; Lin, S.-C.; Lanz, R.B.; Creighton, C.J.; DeMayo, F.; Tsai, S.Y.; Tsai, M.-J. Androgen deprivation–induced NCoA2 promotes metastatic and castration-resistant prostate cancer. J. Clin. Investig. 2014, 124, 5013–5026. [Google Scholar] [CrossRef]
- Silva, M.P.; Barros-Silva, J.D.; Vieira, J.; Lisboa, S.; Torres, L.; Correia, C.; Vieira-Coimbra, M.; Martins, A.T.; Jerónimo, C.; Henrique, R.; et al. NCOA2 is a candidate target gene of 8q gain associated with clinically aggressive prostate cancer. Genes Chromosom. Cancer 2016, 55, 365–374. [Google Scholar] [CrossRef]
- Long, M.D.; Jacobi, J.J.; Singh, P.K.; Llimos, G.; Wani, S.A.; Rowsam, A.M.; Rosario, S.R.; Hoogstraat, M.; Linder, S.; Kirk, J.; et al. Reduced NCOR2 expression accelerates androgen deprivation therapy failure in prostate cancer. Cell Rep. 2021, 37, 110109. [Google Scholar] [CrossRef]
- Long, M.D.; Singh, P.K.; Llimos, G.; Rosario, S.R.; Smiraglia, D.J.; Campbell, M.J. Stage-specific and global functions of NCOR2 in prostate cancer progression. Endocr. Abstr. 2018, 54. [Google Scholar] [CrossRef]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef]
- Adler, D.; Offermann, A.; Braun, M.; Menon, R.; Syring, I.; Nowak, M.; Halbach, R.; Vogel, W.; Ruiz, C.; Zellweger, T.; et al. MED12 overexpression is a frequent event in castration-resistant prostate cancer. Endocr. Relat. Cancer 2014, 21, 663–675. [Google Scholar] [CrossRef]
- Gupta, S.; Shukla, S.; Shukla, M.; MacLennan, G.T.; Fu, P. Deregulation of FOXO3A during prostate cancer progression. Int. J. Oncol. 2009, 34, 1613–1620. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-N. Loss of ETV6 expression results in aggressive prostate cancer progression: Modeling through prostate cancer cell lines. In Proceedings of the Global Summit on Oncology & Cancer, Osaka, Japan, 25–27 May 2017. [Google Scholar] [CrossRef]
- Tsai, Y.-C.; Zeng, T.; Abou-Kheir, W.; Yeh, H.-L.; Yin, J.J.; Lee, Y.-C.; Chen, W.-Y.; Liu, Y.-N. Disruption of ETV6 leads to TWIST1-dependent progression and resistance to epidermal growth factor receptor tyrosine kinase inhibitors in prostate cancer. Mol. Cancer 2018, 17, 42. [Google Scholar] [CrossRef] [PubMed]
- Cosi, I.; Pellecchia, A.; De Lorenzo, E.; Torre, E.; Sica, M.; Nesi, G.; Notaro, R.; De Angioletti, M. ETV4 promotes late development of prostatic intraepithelial neoplasia and cell proliferation through direct and p53-mediated downregulation of p21. J. Hematol. Oncol. 2020, 13, 112. [Google Scholar] [CrossRef] [PubMed]
- Pellecchia, A.; Pescucci, C.; De Lorenzo, E.; Luceri, C.; Passaro, N.; Sica, M.; Notaro, R.; De Angioletti, M. Overexpression of ETV4 is oncogenic in prostate cells through promotion of both cell proliferation and epithelial to mesenchymal transition. Oncogenesis 2012, 1, e20. [Google Scholar] [CrossRef] [PubMed]
- Aytes, A.; Mitrofanova, A.; Kinkade, C.W.; Lefebvre, C.; Lei, M.; Phelan, V.; LeKaye, H.C.; Koutcher, J.A.; Cardiff, R.D.; Califano, A.; et al. ETV4 promotes metastasis in response to activation of PI3-kinase and Ras signaling in a mouse model of advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2013, 110, E3506–E3515. [Google Scholar] [CrossRef] [PubMed]
- Hägglöf, C.; Hammarsten, P.; Strömvall, K.; Egevad, L.; Josefsson, A.; Stattin, P.; Granfors, T.; Bergh, A. TMPRSS2-ERG Expression Predicts Prostate Cancer Survival and Associates with Stromal Biomarkers. PLoS ONE 2014, 9, e86824. [Google Scholar] [CrossRef]
- Baldus, C.D.; Burmeister, T.; Martus, P.; Schwartz, S.; Gökbuget, N.; Bloomfield, C.D.; Hoelzer, D.; Thiel, E.; Hofmann, W.K. High Expression of the ETS Transcription Factor ERG Predicts Adverse Outcome in Acute T-Lymphoblastic Leukemia in Adults. J. Clin. Oncol. 2006, 24, 4714–4720. [Google Scholar] [CrossRef]
- Shimizu, K.; Ichikawa, H.; Tojo, A.; Kaneko, Y.; Maseki, N.; Hayashi, Y.; Ohira, M.; Asano, S.; Ohki, M. An ETS-related gene, ERG, is rearranged in human myeloid leukemia with t(16;21) chromosomal translocation. Proc. Natl. Acad. Sci. USA 1993, 90, 10280–10284. [Google Scholar] [CrossRef]
- Toubaji, A.; Albadine, R.; Meeker, A.K.; Isaacs, W.B.; Lotan, T.; Haffner, M.C.; Chaux, A.; Epstein, I.J.; Han, M.; Walsh, P.C.; et al. Increased gene copy number of ERG on chromosome 21 but not TMPRSS2–ERG fusion predicts outcome in prostatic adenocarcinomas. Mod. Pathol. 2011, 24, 1511–1520. [Google Scholar] [CrossRef]
- Albadine, R.; Latour, M.; Toubaji, A.; Haffner, M.; Isaacs, W.B.; Platz, A.E.; Meeker, A.K.; Demarzo, A.M.; Epstein, I.J.; Netto, G.J. TMPRSS2–ERG gene fusion status in minute (minimal) prostatic adenocarcinoma. Mod. Pathol. 2009, 22, 1415–1422. [Google Scholar] [CrossRef]
- Zong, Y.; Xin, L.; Goldstein, A.S.; Lawson, D.A.; Teitell, M.A.; Witte, O.N. ETS family transcription factors collaborate with alternative signaling pathways to induce carcinoma from adult murine prostate cells. Proc. Natl. Acad. Sci. USA 2009, 106, 12465–12470. [Google Scholar] [CrossRef]
- Kumar-Sinha, C.; Tomlins, S.A.; Chinnaiyan, A.M. Recurrent gene fusions in prostate cancer. Nat. Rev. Cancer 2008, 8, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Perner, S.; Mosquera, J.-M.; Demichelis, F.; Hofer, M.D.; Paris, P.L.; Simko, J.; Collins, C.; Bismar, T.A.; Chinnaiyan, A.M.; De Marzo, A.M.; et al. TMPRSS2-ERG Fusion Prostate Cancer: An Early Molecular Event Associated with Invasion. Am. J. Surg. Pathol. 2007, 31, 882–888. [Google Scholar] [CrossRef]
- Shrestha, E.; Coulter, J.B.; Guzman, W.; Ozbek, B.; Hess, M.M.; Mummert, L.; Ernst, S.E.; Maynard, J.P.; Meeker, A.K.; Heaphy, C.M.; et al. Oncogenic gene fusions in nonneoplastic precursors as evidence that bacterial infection can initiate prostate cancer. Proc. Natl. Acad. Sci. USA 2021, 118, e2018976118. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Zhang, J.; He, X.; Gu, Y.; Qian, B.Z.; Xie, R.; Yu, W.; Zhang, X.; Li, T.; Shi, X.; et al. SPP1 promotes enzalutamide resistance and epithelial-mesenchymal-transition activation in castration-resistant prostate cancer via PI3K/AKT and ERK1/2 Pathways. Oxid. Med. Cell. Longev. 2021, 2021, 5806602. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yu, J.; Wang, D.; Niu, Y.; Chen, S.; Gao, P.; Yang, Z.; Wang, H.; Zhang, J.; Zhang, C.; et al. Epigenetically upregulated microRNA-602 is involved in a negative feedback loop with FOXK2 in esophageal squamous cell carcinoma. Mol. Ther. 2019, 2, 1796–1809. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, N.; Haqqi, T.M. Microrna-453 regulates IL-1β-induced Sonic Hedgehog (SHH) gene expression in osteoarthritis. Osteoarthr. Cartil. 2012, 2, S138–S139. [Google Scholar] [CrossRef]
- Yang, L.; Ma, Z.; Wang, D.; Zhao, W.; Chen, L.; Wang, G. MicroRNA-602 regulating tumor suppressive gene RASSF1A is over-expressed in hepatitis B virus-infected liver and hepatocellular carcinoma. Cancer Biol. Ther. 2010, 9, 803–808. [Google Scholar] [CrossRef]
- Lin, J.; Wang, S.; Shen, H.; Zheng, B. The expression profile of miRNA in glioma and the role of miR-339-5p in glioma. Biomed. Res. Int. 2022, 2022, 4085039. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-N.; Chen, Z.-H.; Chen, W.-C. Novel circulating microRNAs expression profile in colon cancer: A pilot study. Eur. J. Med. Res. 2017, 22, 51. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Gao, K.; Luo, H.; Wang, X.; Shi, Y.; Dong, Q.; Luan, W.; You, Y. Identification of intrinsic subtype-specific prognostic microRNAs in primary glioblastoma. J. Exp. Clin. Cancer Res. 2014, 33, 9. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Zhu, E.C.; Qu, Y.-L.; Wang, D.-Y.; Qu, W.-W.; Zhang, C.-G.; Wu, T.; Gao, Z.-H. Serum level of co-expressed hub miRNAs as diagnostic and prognostic biomarkers for pancreatic ductal adenocarcinoma. J. Cancer 2018, 9, 3991–3999. [Google Scholar] [CrossRef] [PubMed]
- Schnabl, B.; Valletta, D.; Kirovski, G.; Hellerbrand, C. Zinc finger protein 267 is up-regulated in hepatocellular carcinoma and promotes tumor cell proliferation and migration. Exp. Mol. Pathol. 2011, 91, 695–701. [Google Scholar] [CrossRef]
- Lu, L.; Chen, X.; Tao, H.; Xiong, W.; Jie, S.; Li, H. Regulation of the expression of zinc finger protein genes by microRNAs enriched within acute lymphoblastic leukemia-derived microvesicles. Genet. Mol. Res. 2015, 14, 11884–11895. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, L.; Zheng, Y.; Hu, G.; Ma, H.; Shen, L. Knockdown of zinc finger protein 267 suppresses diffuse large B-cell lymphoma progression, metastasis, and cancer stem cell properties. Bioengineered 2022, 13, 1686–1701. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Meyers, T.J.; Emeka, A.A.; Cooley, L.F.; Cooper, P.R.; Lancki, N.; Helenowski, I.; Kachuri, L.; Lin, D.W.; Stanford, J.L.; et al. Genetic factors associated with prostate cancer conversion from active surveillance to treatment. Hum. Genet. Genom. Adv. 2022, 3, 100070. [Google Scholar] [CrossRef]
- Zhang, C.Q.; Williams, H.; Prince, T.L.; Ho, E.S. Overexpressed HSF1 cancer signature genes cluster in human chromosome 8q. Hum. Genom. 2017, 11, 35. [Google Scholar] [CrossRef]
- Sharbatoghli, M.; Fattahi, F.; Es, H.A.; Akbari, A.; Akhavan, S.; Ebrahimi, M.; Asadi-Lari, M.; Totonchi, M.; Madjd, Z. Copy Number Variation of Circulating Tumor DNA (ctDNA) Detected Using NIPT in Neoadjuvant Chemotherapy-Treated Ovarian Cancer Patients. Front. Genet. 2022, 13, 938985. [Google Scholar] [CrossRef]
- Harada, K.; Song, S.; Xu, Y.; Bagewell, B.D.; Pizzi, M.P.; Jin, J.; Wang, Y.; Scott, A.W.; Ma, L.; Amlashi, F.G.; et al. Abstract 288: Genomic profiling of metastatic gastric adenocarcinoma. Cancer Res 2018, 78, 288. [Google Scholar] [CrossRef]
- Richard, I.A.; Burgess, J.T.; O’Byrne, K.J.; Bolderson, E. Beyond PARP1: The potential of other members of the poly (ADP-ribose) polymerase family in DNA repair and cancer therapeutics. Front. Cell Dev. Biol. 2022, 9, 801200. [Google Scholar] [CrossRef]
- Challa, S.; Stokes, M.S.; Kraus, W.L. MARTs and MARylation in the cytosol: Biological functions, mechanisms of action, and therapeutic potential. Cells 2021, 10, 313. [Google Scholar] [CrossRef] [PubMed]
- Nizi, M.G.; Maksimainen, M.M.; Lehtiö, L.; Tabarrini, O. Medicinal Chemistry Perspective on Targeting Mono-ADP-Ribosylating PARPs with Small Molecules. J. Med. Chem. 2022, 65, 7532–7560. [Google Scholar] [CrossRef] [PubMed]
- Mok, K.; Tsoi, H.; Man, E.P.; Leung, M.; Chau, K.M.; Wong, L.; Chan, W.; Chan, S.; Luk, M.; Chan, J.Y.; et al. Repurposing hyperpolarization-activated cyclic nucleotide-gated channels as a novel therapy for breast cancer. Clin. Transl. Med. 2021, 11, e578. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.H.; Yang, S.Y.; You, S.L.; Lien, H.C.; Lin, C.H.; Lin, P.H.; Huang, C.S. Polymorphisms of ESR1, UGT1A1, HCN1, MAP3K1 and CYP2B6 are associated with the prognosis of hormone receptor-positive early breast cancer. Oncotarget 2017, 8, 20925. [Google Scholar] [CrossRef]
- Phan, N.N.; Huynh, T.T.; Lin, Y.-C. Hyperpolarization-activated cyclic nucleotide-gated gene signatures and poor clinical outcome of cancer patient. Transl. Cancer Res. 2017, 6, 698–708. [Google Scholar] [CrossRef]


{kind=link}
| Clinical and Molecular Features | HR * | 95.0% C.I. * HR | p Value |
|---|---|---|---|
| Disease stage | 4.002 | 2.117–7.565 | 1.97 × 10−5 |
| Prognostic group grade | 0.609 | 0.401–0.926 | 2.05 × 10−2 |
| MIR602 deletion | 2.795 | 1.044–7.484 | 4.09 × 10−2 |
| MIR602 amplification | 1.917 | 1.089–3.376 | 2.42 × 10−2 |
| PARP8 deletion | 1.724 | 1.024–2.904 | 4.05 × 10−2 |
| ZNF267 deletion | 2.256 | 1.280–3.975 | 4.88 × 10−2 |
| MROH1 deletion | 2.519 | 1.116–5.687 | 2.62 × 10−2 |
| HCN1 deletion | 1.774 | 1.056–2.981 | 3.03 × 10−2 |
| Predictors | B | p Value | Exp(B) | 95% C.I. Exp(B) |
|---|---|---|---|---|
| SPOP alteration | 1.554 | 0.014 | 4.728 | 1.362–16.414 |
| SPP1 alteration | 2.583 | 0.013 | 13.231 | 1.716–102.024 |
| CCND1 amplification | 1.425 | 0.006 | 4.157 | 1.500–11.522 |
| CDKN1B deletion | 0.762 | 0.007 | 2.143 | 1.228–3.740 |
| NKX3.1 deletion | 0.715 | 0.001 | 2.045 | 1.341–3.118 |
| PTEN deletion | 0.709 | 0.004 | 2.032 | 1.256–3.287 |
| PARP8 deletion | 0.978 | 0.029 | 2.658 | 1.106–6.386 |
| Constant | −0.624 | <0.001 | 0.536 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfahed, A.; Ebili, H.O.; Almoammar, N.E.; Alasiri, G.; AlKhamees, O.A.; Aldali, J.A.; Al Othaim, A.; Hakami, Z.H.; Abdulwahed, A.M.; Waggiallah, H.A. Prognostic Values of Gene Copy Number Alterations in Prostate Cancer. Genes 2023, 14, 956. https://doi.org/10.3390/genes14050956
Alfahed A, Ebili HO, Almoammar NE, Alasiri G, AlKhamees OA, Aldali JA, Al Othaim A, Hakami ZH, Abdulwahed AM, Waggiallah HA. Prognostic Values of Gene Copy Number Alterations in Prostate Cancer. Genes. 2023; 14(5):956. https://doi.org/10.3390/genes14050956
Chicago/Turabian StyleAlfahed, Abdulaziz, Henry Okuchukwu Ebili, Nasser Eissa Almoammar, Glowi Alasiri, Osama A. AlKhamees, Jehad A. Aldali, Ayoub Al Othaim, Zaki H. Hakami, Abdulhadi M. Abdulwahed, and Hisham Ali Waggiallah. 2023. "Prognostic Values of Gene Copy Number Alterations in Prostate Cancer" Genes 14, no. 5: 956. https://doi.org/10.3390/genes14050956