
Metabolic Myopathies in the Era of Next-Generation Sequencing



Abstract
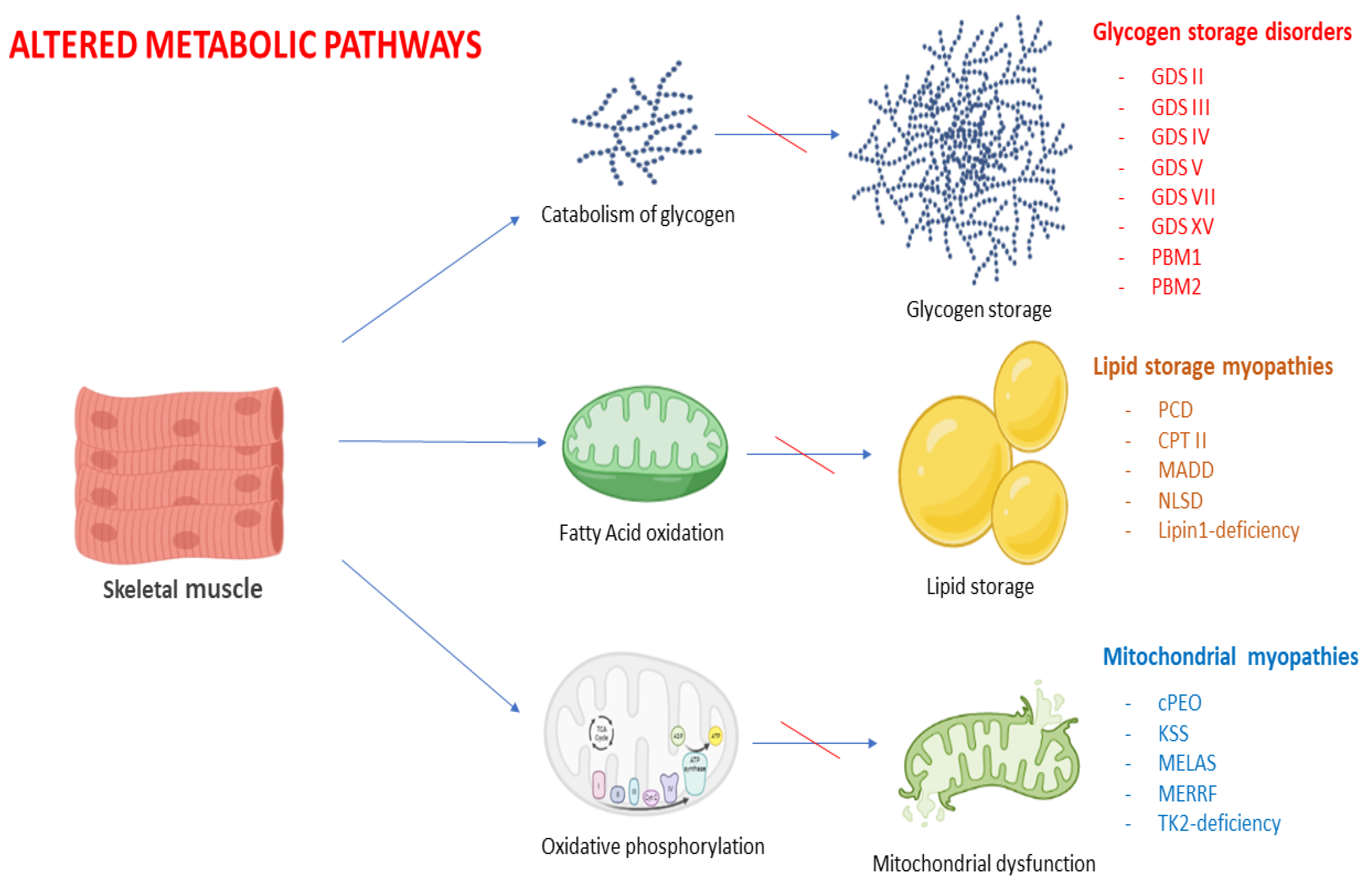
:1. Introduction
2. General Considerations
2.1. Overview of Muscle Metabolism
2.2. Epidemiology
2.3. NGS in Metabolic Myopathies: Advantages and Challenges
2.4. Clinical Presentation
2.5. Diagnostic Tools
2.6. Diagnostic Algorithms
2.7. Therapeutics
3. Glycogen Storage Disorders
3.1. Pompe Disease
3.2. McArdle Disease
3.3. Other Muscle Glycogenoses
3.4. Polyglucosan Body Myopathies
4. Lipid Myopathies
4.1. Fatty Acid Oxidation Disorders
4.1.1. Primary Carnitine Deficiency
4.1.2. Multiple Acyl-CoA Dehydrogenase Deficiency
4.1.3. Carnitine Palmitoyl Transferase II Deficiency
4.1.4. Neutral Lipid Storage Diseases
5. Primary Mitochondrial Myopathies
5.1. TK2 Deficiency
5.2. Lipin-1 Deficiency
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tarnopolsky, M.A. Metabolic Myopathies. Contin. Lifelong Learn. Neurol. 2016, 22, 1829–1851. [Google Scholar] [CrossRef]
- Angelini, C.; Marozzo, R.; Pegoraro, V.; Sacconi, S. Diagnostic challenges in metabolic myopathies. Expert Rev. Neurother. 2020, 20, 1287–1298. [Google Scholar] [CrossRef]
- Parikh, S.; Goldstein, A.; Koenig, M.K.; Scaglia, F.; Enns, G.M.; Saneto, R.; Anselm, I.; Cohen, B.H.; Falk, M.J.; Greene, C.; et al. Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Anesthesia Analg. 2014, 17, 689–701. [Google Scholar] [CrossRef]
- Guémy, C.; Laforêt, P. The new horizons for treatment of Late-Onset Pompe Disease (LOPD). Rev. Neurol. 2023, 179, 81–89. [Google Scholar] [CrossRef]
- Byrne, B.; Bratkovic, D.; Díaz-Manera, J.; Laforêt, P.; Mozaffar, T.; van der Ploeg, A.; Roberts, M.; Schoser, B.; Toscano, A.; Jiang, H.; et al. Cipaglucosidase alfa/miglustat versus alglucosidase alfa/placebo in late-onset Pompe disease (LOPD): PROPEL study subgroup analyses. Mol. Genet. Metab. 2022, 135, S27–S28. [Google Scholar] [CrossRef]
- Sawada, T.; Kido, J.; Nakamura, K. Newborn Screening for Pompe Disease. Int. J. Neonatal Screen. 2020, 6, 31. [Google Scholar] [CrossRef]
- Glancy, B.; Balaban, R.S. Energy metabolism design of the striated muscle cell. Physiol. Rev. 2021, 101, 1561–1607. [Google Scholar] [CrossRef]
- Al Shehri, A.; Al-Asmi, A.; Al Salti, A.M.; Almadani, A.; Hassan, A.; Bamaga, A.K.; Cupler, E.J.; Al-Hashel, J.; Alabdali, M.M.; Alanazy, M.H.; et al. A Multidisciplinary Perspective Addressing the Diagnostic Challenges of Late-Onset Pompe Disease in the Arabian Peninsula Region Developed from an Expert Group Meeting. J. Neuromuscul. Dis. 2022, 9, 661–673. [Google Scholar] [CrossRef]
- Elenga, N.; Verloes, A.; Mrsic, Y.; Basurko, C.; Schaub, R.; Cuadro-Alvarez, E.; Kom-Tchameni, R.; Carles, G.; Lambert, V.; Boukhari, R.; et al. Incidence of infantile Pompe disease in the Maroon population of French Guiana. BMJ Paediatr. Open 2018, 2, e000182. [Google Scholar] [CrossRef]
- Pizzamiglio, C.; Mahroo, O.A.; Khan, K.N.; Patasin, M.; Quinlivan, R. Phenotype and genotype of 197 British patients with McArdle disease: An observational single-centre study. J. Inherit. Metab. Dis. 2021, 44, 1409–1418. [Google Scholar] [CrossRef]
- Nilipour, Y.; Fatehi, F.; Sanatinia, S.; Bradshaw, A.; Duff, J.; Lochmüller, H.; Horvath, R.; Nafissi, S. Multiple acyl-coenzyme A dehydrogenase deficiency shows a possible founder effect and is the most frequent cause of lipid storage myopathy in Iran. J. Neurol. Sci. 2020, 411, 116707. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-González, C.; Madruga-Garrido, M.; Hirano, M.; Martí, I.; Martín, M.A.; Munell, F.; Nascimento, A.; Olivé, M.; Quan, J.; Sardina, M.D.; et al. Collaborative model for diagnosis and treatment of very rare diseases: Experience in Spain with thymidine kinase 2 deficiency. Orphanet J. Rare Dis. 2021, 16, 407. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.W.P.; Chin, H.-L.; Chin, A.X.Y.; Goh, D.L.-M. Using gene panels in the diagnosis of neuromuscular disorders: A mini-review. Front. Neurol. 2022, 13, 997551. [Google Scholar] [CrossRef]
- Benarroch, L.; Bonne, G.; Rivier, F.; Hamroun, D. The 2023 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul. Disord. 2022, 33, 76–117. [Google Scholar] [CrossRef]
- Kerr, M.; Hume, S.; Omar, F.; Koo, D.; Barnes, H.; Khan, M.; Aman, S.; Wei, X.-C.; Alfuhaid, H.; McDonald, R.; et al. MITO-FIND: A study in 390 patients to determine a diagnostic strategy for mitochondrial disease. Mol. Genet. Metab. 2020, 131, 66–82. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Anesthesia Analg. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Harrison, S.M.; Rehm, H.L. Is ‘likely pathogenic’ really 90% likely? Reclassification data in ClinVar. Genome Med. 2019, 11, 72. [Google Scholar] [CrossRef]
- Ravaglia, S.; Malovini, A.; Cirio, S.; Danesino, C.; De Filippi, P.; Moggio, M.; Mongini, T.; Maggi, L.; Servidei, S.; Vianello, A.; et al. Polymorphism in exercise genes and respiratory function in late-onset Pompe disease. J. Appl. Physiol. 2021, 131, 1762–1771. [Google Scholar] [CrossRef] [PubMed]
- Toscano, A.; Barca, E.; Musumeci, O. Update on diagnostics of metabolic myopathies. Curr. Opin. Neurol. 2017, 30, 553–562. [Google Scholar] [CrossRef]
- Haller, R.G.; Vissing, J. No spontaneous second wind in muscle phosphofructokinase deficiency. Neurology 2004, 62, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, E.M.; Garibaldi, M.; Antonini, G. Lipid Myopathies. J. Clin. Med. 2018, 7, 472. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef]
- Zutt, R.; van der Kooi, A.; Linthorst, G.; Wanders, R.; de Visser, M. Rhabdomyolysis: Review of the literature. Neuromuscul. Disord. 2014, 24, 651–659. [Google Scholar] [CrossRef]
- Kruijt, N.; Bersselaar, L.V.D.; Kamsteeg, E.J.; Verbeeck, W.; Snoeck, M.M.J.; Everaerd, D.S.; Abdo, W.F.; Jansen, D.R.M.; Erasmus, C.E.; Jungbluth, H.; et al. The etiology of rhabdomyolysis: An interaction between genetic susceptibility and external triggers. Eur. J. Neurol. 2021, 28, 647–659. [Google Scholar] [CrossRef]
- Gemelli, C.; Traverso, M.; Trevisan, L.; Fabbri, S.; Scarsi, E.; Carlini, B.; Prada, V.; Mongini, T.; Ruggiero, L.; Patrone, S.; et al. An integrated approach to the evaluation of patients with asymptomatic or minimally symptomatic hyperCKemia. Muscle Nerve 2022, 65, 96–104. [Google Scholar] [CrossRef]
- Mbbs, A.V.; Shashi, V.; Jiang, Y.-H.; Do, W.B.G.; Schoch, K.; Smith, E.C. Pseudometabolic presentation of dystrophinopathy due to a missense mutation. Muscle Nerve 2010, 42, 975–979. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Adesina, A.M.; Jones, J.; Scaglia, F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metab. 2015, 116, 4–12. [Google Scholar] [CrossRef]
- Hogrel, J.-Y.; Laforet, P.; Ben Yaou, R.; Chevrot, M.; Eymard, B.; Lombes, A. A non-ischemic forearm exercise test for the screening of patients with exercise intolerance. Neurology 2001, 56, 1733–1738. [Google Scholar] [CrossRef]
- Malfatti, E.; Romero, N.B. Diseases of the skeletal muscle. Handb. Clin. Neurol. 2018, 145, 429–451. [Google Scholar] [CrossRef]
- Hwang, J.-H.; Choi, C.S. Use of in vivo magnetic resonance spectroscopy for studying metabolic diseases. Exp. Mol. Med. 2015, 47, e139. [Google Scholar] [CrossRef]
- Goldstein, A.; Falk, M.J. Mitochondrial DNA Deletion Syndromes. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2003; updated 31 January 2019. [Google Scholar]
- Preisler, N.; Orngreen, M.C.; Echaniz-Laguna, A.; Laforet, P.; Lonsdorfer-Wolf, E.; Doutreleau, S.; Geny, B.; Akman, H.O.; DiMauro, S.; Vissing, J. Muscle phosphorylase kinase deficiency: A neutral metabolic variant or a disease? Neurology 2012, 78, 265–268. [Google Scholar] [CrossRef]
- Angel, A.G.-D.; Bisciglia, M.; Vargas-Cañas, S.; Fernandez-Valverde, F.; Kazakova, E.; Escobar, R.E.; Romero, N.B.; Jardel, C.; Rucheton, B.; Stojkovic, T.; et al. Novel Phenotypes and Cardiac Involvement Associated with DNA2 Genetic Variants. Front. Neurol. 2019, 10, 1049. [Google Scholar] [CrossRef] [PubMed]
- Bersselaar, L.R.V.D.; Jungbluth, H.; Kruijt, N.; Kamsteeg, E.-J.; A. Fernandez-Garcia, M.; Treves, S.; Riazi, S.; Malagon, I.; van Eijk, L.T.; van Alfen, N.; et al. Neuromuscular symptoms in patients with RYR1-related malignant hyperthermia and rhabdomyolysis. Brain Commun. 2022, 4, fcac292. [Google Scholar] [CrossRef] [PubMed]
- Beam, T.A.; Loudermilk, E.F.; Kisor, D.F. Pharmacogenetics and pathophysiology of CACNA1S mutations in malignant hyperthermia. Physiol. Genom. 2017, 49, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Tarnopolsky, M.A.; Parise, G.; Gibala, M.J.; Graham, T.E.; Rush, J.W.E. Myoadenylate deaminase deficiency does not affect muscle anaplerosis during exhaustive exercise in humans. J. Physiol. 2001, 533, 881–889. [Google Scholar] [CrossRef]
- Cakmak, E.; Bagci, G. Chanarin-Dorfman Syndrome: A comprehensive review. Liver Int. 2021, 41, 905–914. [Google Scholar] [CrossRef]
- Madsen, K.L.; Laforêt, P.; Buch, A.E.; Stemmerik, M.G.; Ottolenghi, C.; Hatem, S.N.; Raaschou-Pedersen, D.T.; Poulsen, N.S.; Atencio, M.; Luton, M.; et al. No effect of triheptanoin on exercise performance in McArdle disease. Ann. Clin. Transl. Neurol. 2019, 6, 1949–1960. [Google Scholar] [CrossRef]
- Domínguez-González, C.; Madruga-Garrido, M.; Mavillard, F.; Garone, C.; Aguirre-Rodríguez, F.J.; Donati, M.A.; Kleinsteuber, K.; Martí, I.; Martín-Hernández, E.; Morealejo-Aycinena, J.P.; et al. Deoxynucleoside Therapy for Thymidine Kinase 2–Deficient Myopathy. Ann. Neurol. 2019, 86, 293–303. [Google Scholar] [CrossRef]
- Vissing, J.; Haller, R.G. The Effect of Oral Sucrose on Exercise Tolerance in Patients with McArdle’s Disease. New Engl. J. Med. 2003, 349, 2503–2509. [Google Scholar] [CrossRef]
- Barp, A.; Bellance, R.; Malfatti, E.; Rigal, O.; Acquaviva-Bourdain, C.; Laforet, P. Late Onset Multiple Acyl-CoA Dehydrogenase Deficiency (MADD) Myopathy Misdiagnosed as Polymyositis. Am. J. Clin. Oncol. 2019, 26, e125–e127. [Google Scholar] [CrossRef]
- Ørngreen, M.C.; Madsen, K.L.; Preisler, N.; Andersen, G.; Vissing, J.; Laforêt, P. Bezafibrate in skeletal muscle fatty acid oxidation disorders: A randomized clinical trial. Neurology 2014, 82, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Meena, N.K.; Raben, N. Pompe Disease: New Developments in an Old Lysosomal Storage Disorder. Biomolecules 2020, 10, 1339. [Google Scholar] [CrossRef] [PubMed]
- Preisler, N.; Laforet, P.; Madsen, K.L.; Hansen, R.S.; Lukacs, Z.; Ørngreen, M.C.; Lacour, A.; Vissing, J. Fat and carbohydrate metabolism during exercise in late-onset Pompe disease. Mol. Genet. Metab. 2012, 107, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Young, S.P.; Bs, M.C.; Bs, G.H.D.; Zhang, H.; Dai, J.; Bs, D.P.; Millington, D.S.; Kishnani, P.S.; Bali, D.S. Screening for pompe disease using a rapid dried blood spot method: Experience of a clinical diagnostic laboratory. Muscle Nerve 2009, 40, 32–36. [Google Scholar] [CrossRef]
- Reuser, A.J.J.; van der Ploeg, A.T.; Chien, Y.; Llerena, J.; Abbott, M.; Clemens, P.R.; Kimonis, V.E.; Leslie, N.; Maruti, S.S.; Sanson, B.; et al. GAA variants and phenotypes among 1,079 patients with Pompe disease: Data from the Pompe Registry. Hum. Mutat. 2019, 40, 2146–2164. [Google Scholar] [CrossRef]
- Shemesh, A.; Wang, Y.; Yang, Y.; Yang, G.-S.; Johnson, D.E.; Backer, J.M.; Pessin, J.E.; Zong, H. Suppression of mTORC1 activation in acid-α-glucosidase-deficient cells and mice is ameliorated by leucine supplementation. Am. J. Physiol. Integr. Comp. Physiol. 2014, 307, R1251–R1259. [Google Scholar] [CrossRef]
- Tarnopolsky, M.A.; Nilsson, M.I. Nutrition and exercise in Pompe disease. Ann. Transl. Med. 2019, 7, 282. [Google Scholar] [CrossRef]
- Sheikh, A.M.; Vissing, J. Exercise therapy for muscle and lower motor neuron diseases. Acta Myol. 2019, 38, 215–232. [Google Scholar]
- Scalco, R.S.; EUROMAC Consortium; Lucia, A.; Santalla, A.; Martinuzzi, A.; Vavla, M.; Reni, G.; Toscano, A.; Musumeci, O.; Voermans, N.C.; et al. Data from the European registry for patients with McArdle disease and other muscle glycogenoses (EUROMAC). Orphanet J. Rare Dis. 2020, 15, 330. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Lornage, X.; Laforêt, P.; Orngreen, M.C.; Edelweiss, E.; Brochier, G.; Bui, M.T.; Silva-Rojas, R.; Birck, C.; Lannes, B.; et al. A New Glycogen Storage Disease Caused by a Dominant PYGM Mutation. Ann. Neurol. 2020, 88, 274–282. [Google Scholar] [CrossRef]
- Toscano, A.; Musumeci, O. Tarui disease and distal glycogenoses: Clinical and genetic update. Acta Myol. Myopathies Cardiomyopathies J. Mediterr. Soc. Myol. 2007, 26, 105–107. [Google Scholar]
- Andersen, S.T.; Vissing, J. Carbohydrate- and protein-rich diets in McArdle disease: Effects on exercise capacity. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1359–1363. [Google Scholar] [CrossRef]
- Løkken, N.; Voermans, N.C.; Andersen, L.K.; Karazi, W.; Reason, S.L.; Zweers, H.; Wilms, G.; Santalla, A.; Susanibar, E.; Lucia, A.; et al. Patient-Reported Experiences with a Low-Carbohydrate Ketogenic Diet: An International Survey in Patients with McArdle Disease. Nutrients 2023, 15, 843. [Google Scholar] [CrossRef] [PubMed]
- Quinlivan, R.; Martinuzzi, A.; Schoser, B. Pharmacological and nutritional treatment for McArdle disease (Glycogen Storage Disease type V). Cochrane Database Syst. Rev. 2014, 2014, CD003458. [Google Scholar] [CrossRef] [PubMed]
- Decostre, V.; Laforêt, P.; De Antonio, M.; Kachetel, K.; Canal, A.; Ollivier, G.; Nadaj-Pakleza, A.; Petit, F.M.; Wahbi, K.; Fayssoil, A.; et al. Long term longitudinal study of muscle function in patients with glycogen storage disease type IIIa. Mol. Genet. Metab. 2017, 122, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Hoogeveen, I.J.; van der Ende, R.M.; van Spronsen, F.J.; de Boer, F.; Heiner-Fokkema, M.R.; Derks, T.G.J. Normoglycemic Ketonemia as Biochemical Presentation in Ketotic Glycogen Storage Disease. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2015; Volume 28, pp. 41–47. [Google Scholar] [CrossRef]
- Schreuder, A.B.; Rossi, A.; Grünert, S.C.; Derks, T.G.J. Glycogen Storage Disease Type III. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2010; updated 6 January 2022. [Google Scholar]
- Laforêt, P.; Inoue, M.; Goillot, E.; Lefeuvre, C.; Cagin, U.; Streichenberger, N.; Leonard-Louis, S.; Brochier, G.; Madelaine, A.; Labasse, C.; et al. Deep morphological analysis of muscle biopsies from type III glycogenesis (GSDIII), debranching enzyme deficiency, revealed stereotyped vacuolar myopathy and autophagy impairment. Acta Neuropathol. Commun. 2019, 7, 167. [Google Scholar] [CrossRef]
- Bao, Y.; Kishnani, P.; Wu, J.Y.; Chen, Y.T. Hepatic and neuromuscular forms of glycogen storage disease type IV caused by mutations in the same glycogen-branching enzyme gene. J. Clin. Investig. 1996, 97, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Assereto, S.; van Diggelen, O.P.; Diogo, L.; Morava, E.; Cassandrini, D.; Carreira, I.; de Boode, W.-P.; Dilling, J.; Garcia, P.; Henriques, M.; et al. Null mutations and lethal congenital form of glycogen storage disease type IV. Biochem. Biophys. Res. Commun. 2007, 361, 445–450. [Google Scholar] [CrossRef]
- Reusche, E.; Aksu, F.; Goebel, H.H.; Shin, Y.S.; Yokota, T.; Reichmann, H. A mild juvenile variant of type IV glycogenosis. Brain Dev. 1992, 14, 36–43. [Google Scholar] [CrossRef]
- Malfatti, E.; Barnerias, C.; Hedberg-Oldfors, C.; Gitiaux, C.; Benezit, A.; Oldfors, A.; Carlier, R.-Y.; Quijano-Roy, S.; Romero, N.B. A novel neuromuscular form of glycogen storage disease type IV with arthrogryposis, spinal stiffness and rare polyglucosan bodies in muscle. Neuromuscul. Disord. 2016, 26, 681–687. [Google Scholar] [CrossRef]
- Laforêt, P.; Malfatti, E.; Vissing, J. Update on new muscle glycogenosis. Curr. Opin. Neurol. 2017, 30, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Laforêt, P.; Oldfors, A.; Malfatti, E.; Vissing, J.; Colle, M.-A.; Duran, J.; Gentry, M.; Guinovart, J.; Hurley, T.; Kakhlon, O.; et al. 251st ENMC international workshop: Polyglucosan storage myopathies 13–15 December 2019, Hoofddorp, The Netherlands. Neuromuscul. Disord. 2021, 31, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Cenacchi, G.; Papa, V.; Costa, R.; Pegoraro, V.; Marozzo, R.; Fanin, M.; Angelini, C. Update on polyglucosan storage diseases. Virchows Arch. 2019, 475, 671–686. [Google Scholar] [CrossRef] [PubMed]
- Lilleker, J.; Keh, Y.S.; Roncaroli, F.; Sharma, R.; Roberts, M. Metabolic myopathies: A practical approach. Prac. Neurol. 2017, 18, 14–26. [Google Scholar] [CrossRef]
- Berardo, A.; DiMauro, S.; Hirano, M. A Diagnostic Algorithm for Metabolic Myopathies. Curr. Neurol. Neurosci. Rep. 2010, 10, 118–126. [Google Scholar] [CrossRef]
- Crefcoeur, L.L.; Visser, G.; Ferdinandusse, S.; Wijburg, F.A.; Langeveld, M.; Sjouke, B. Clinical characteristics of primary carnitine deficiency: A structured review using a case-by-case approach. J. Inherit. Metab. Dis. 2022, 45, 386–405. [Google Scholar] [CrossRef] [PubMed]
- Tobon, A. Metabolic Myopathies. Contin. Lifelong Learn. Neurol. 2013, 19, 1571–1597. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Kumar, K.; Malhotra, S.; Sibal, A. Rare case of primary carnitine deficiency presenting as acute liver failure. BMJ Case Rep. 2022, 15, e247225. [Google Scholar] [CrossRef]
- Mutlu-Albayrak, H.; Bene, J.; Oflaz, M.B.; Tanyalçın, T.; Çaksen, H.; Melegh, B. Identification ofSLC22A5Gene Mutation in a Family with Carnitine Uptake Defect. Case Rep. Genet. 2015, 2015, 259627. [Google Scholar] [CrossRef]
- Gempel, K.; Topaloglu, H.; Talim, B.; Schneiderat, P.; Schoser, B.; Hans, V.H.; Pálmafy, B.; Kale, G.; Tokatli, A.; Quinzii, C.; et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007, 130, 2037–2044. [Google Scholar] [CrossRef]
- Prasun, P. Multiple Acyl-CoA Dehydrogenase Deficiency. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2020. [Google Scholar]
- Tian, Y.; Wang, S.; Wang, F.; Yi, L.; Dong, M.; Huang, X. Late onset of neutral lipid storage disease due to a rare PNPLA2 mutation in a patient with myopathy and cardiomyopathy. Chin. Med. J. 2022, 135, 2389–2391. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; The Mitochondrial Medicine Society; Saneto, R.; Falk, M.J.; Anselm, I.; Cohen, B.H.; Haas, R. A modern approach to the treatment of mitochondrial disease. Curr. Treat. Options Neurol. 2009, 11, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Camp, K.M.; Krotoski, D.; Parisi, M.A.; Gwinn, K.A.; Cohen, B.H.; Cox, C.S.; Enns, G.M.; Falk, M.J.; Goldstein, A.C.; Gopal-Srivastava, R.; et al. Nutritional interventions in primary mitochondrial disorders: Developing an evidence base. Mol. Genet. Metab. 2016, 119, 187–206. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, T.D.; Schwartz, M.; Olsen, D.B.; Wibrand, F.; Krag, T.; Dunø, M.; Hauerslev, S.; Vissing, J. Aerobic training is safe and improves exercise capacity in patients with mitochondrial myopathy. Brain 2006, 129, 3402–3412. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-González, C.; Hernández-Laín, A.; Rivas, E.; Hernández-Voth, A.; Catalán, J.S.; Fernández-Torrón, R.; Fuiza-Luces, C.; García, J.G.; Morís, G.; Olivé, M.; et al. Late-onset thymidine kinase 2 deficiency: A review of 18 cases. Orphanet J. Rare Dis. 2019, 14, 100. [Google Scholar] [CrossRef] [PubMed]
- Zeharia, A.; Shaag, A.; Houtkooper, R.H.; Hindi, T.; de Lonlay, P.; Erez, G.; Hubert, L.; Saada, A.; de Keyzer, Y.; Eshel, G.; et al. Mutations in LPIN1 Cause Recurrent Acute Myoglobinuria in Childhood. Am. J. Hum. Genet. 2008, 83, 489–494. [Google Scholar] [CrossRef]
- Tuchmann-Durand, C.; Roda, C.; Renard, P.; Mortamet, G.; Bérat, C.; Altenburger, L.; de Larauz, M.H.; Thevenet, E.; Cottart, C.; Moulin, F.; et al. Systemic corticosteroids for the treatment of acute episodes of rhabdomyolysis in lipin-1-deficient patients. J. Inherit. Metab. Dis. 2023. ahead of print. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GLYCOGEN STORAGE DISORDERS | LIPID STORAGE MYOPATHIES | MITOCHONDRIAL MYOPATHIES | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GSDII | GSDIII | GSDIV | GSDV | GSDVII | GSDXV | PBM1 * | PBM2 * | PCD | CPT II | MADD | NLSD | Lipin-1 Def. | cPEO | Kearn-Sayre | MELAS | MERRF | TK2 Def. | |
| Muscle phenotype | ||||||||||||||||||
| Ptosis | x | x | x | x | x | |||||||||||||
| Ophtalmoplegia | x | x | x | x | x | |||||||||||||
| Macroglossia | x | |||||||||||||||||
| Myalgia | x | x | x | x | x | x | x | x | x | |||||||||
| Proximal muscle weakness | x | x | x | x | x | x | x | x | x | x | x | x | ||||||
| Distal muscle weakness | x | x | ||||||||||||||||
| Transient muscle weakness | x | |||||||||||||||||
| Rhabdomyolysis | x | x | x | x | x | x | x | x | x | x | ||||||||
| Exercise-induced events | ||||||||||||||||||
| Second-wind phenomenon | x | |||||||||||||||||
| Out-of-wind phenomenon | x | |||||||||||||||||
| Exercise intolerance | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | |||
| Extramuscular signs | ||||||||||||||||||
| CNS | x | x | x | x | x | x | ||||||||||||
| Liver | x | x | x | x | x | |||||||||||||
| Heart | x | x | x | x | x | x | x | x | x | x | ||||||||
| Respiratory | x | x | ||||||||||||||||
| Peripheral neuropathy | x | x | x | |||||||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urtizberea, J.A.; Severa, G.; Malfatti, E. Metabolic Myopathies in the Era of Next-Generation Sequencing. Genes 2023, 14, 954. https://doi.org/10.3390/genes14050954
Urtizberea JA, Severa G, Malfatti E. Metabolic Myopathies in the Era of Next-Generation Sequencing. Genes. 2023; 14(5):954. https://doi.org/10.3390/genes14050954
Chicago/Turabian StyleUrtizberea, Jon Andoni, Gianmarco Severa, and Edoardo Malfatti. 2023. "Metabolic Myopathies in the Era of Next-Generation Sequencing" Genes 14, no. 5: 954. https://doi.org/10.3390/genes14050954