
Generation of an Open-Access Patient-Derived iPSC Biobank for Amyotrophic Lateral Sclerosis Disease Modelling

Abstract
:1. Introduction
2. Materials and Methods
2.1. LCL and PBL Culture
2.2. Generation and Maintenance of iPSCs
2.3. iPSC Characterisation
2.4. Embryoid Body Assay
2.5. Motor Neuron Differentiation
2.6. Immunocytochemistry
2.7. Imaging and Analysis
2.8. Calcium Imaging
3. Results
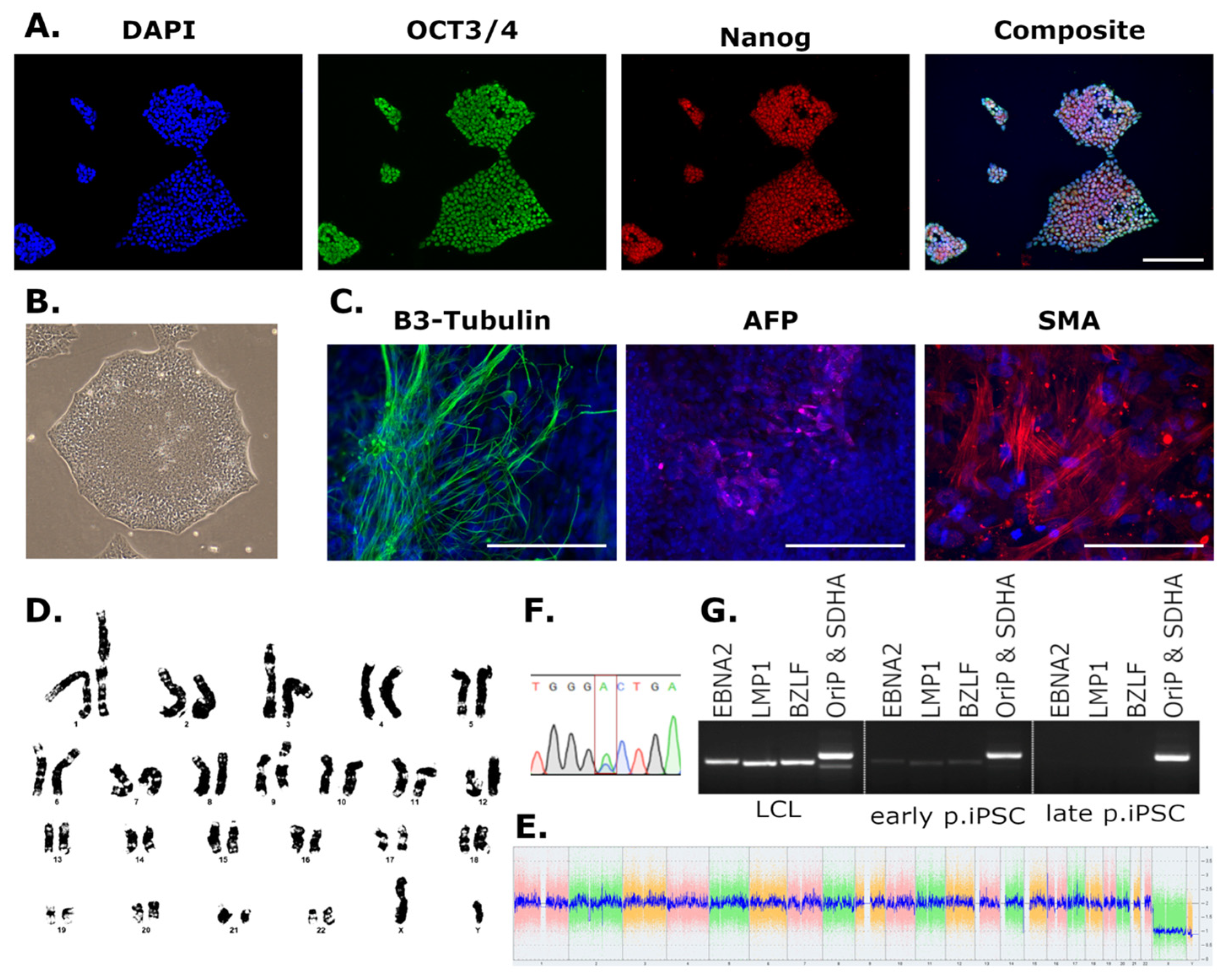
3.1. Generation of iPSCs from LCLs and PBLs
3.2. Characterisation of Newly Derived iPSCs
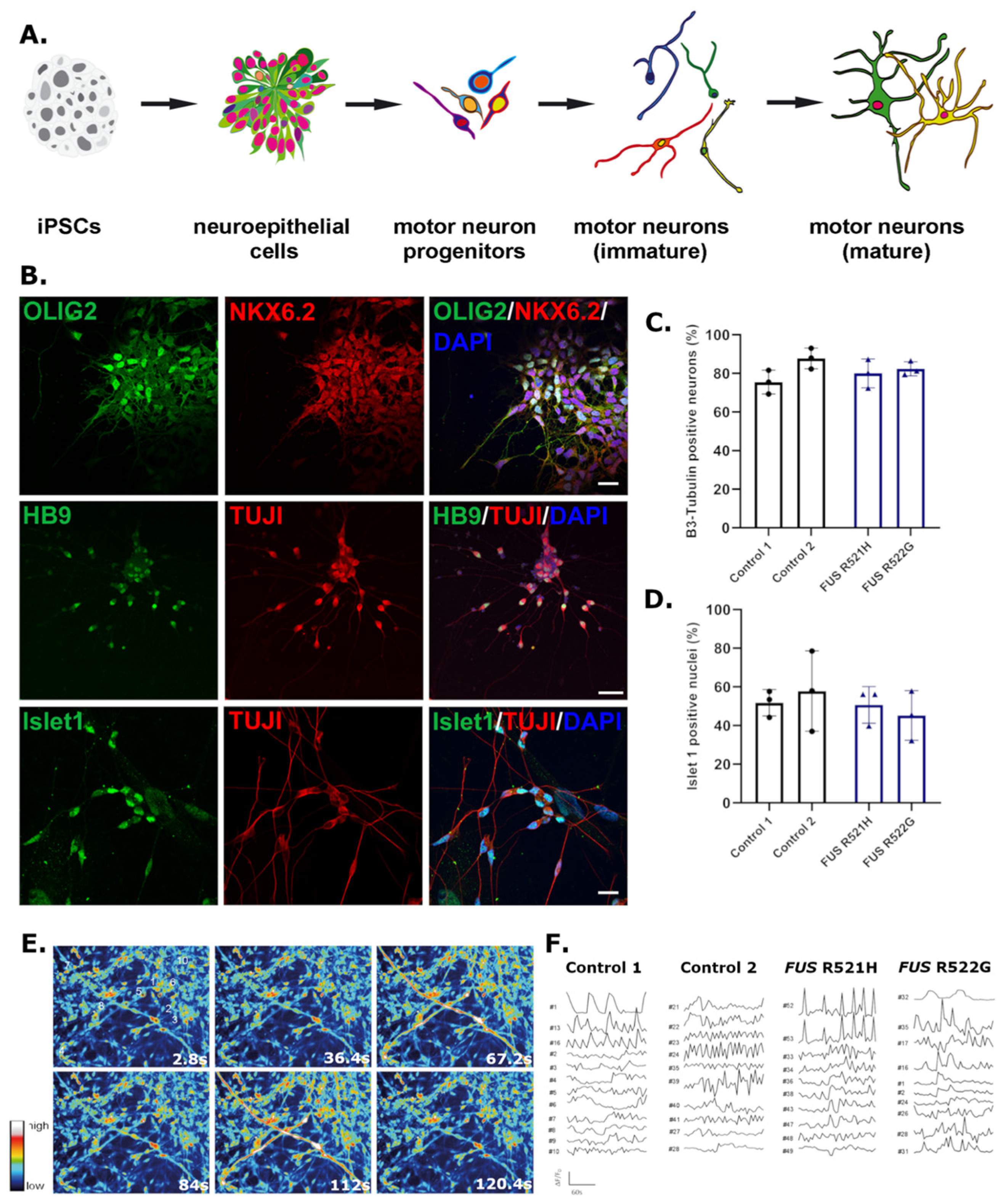
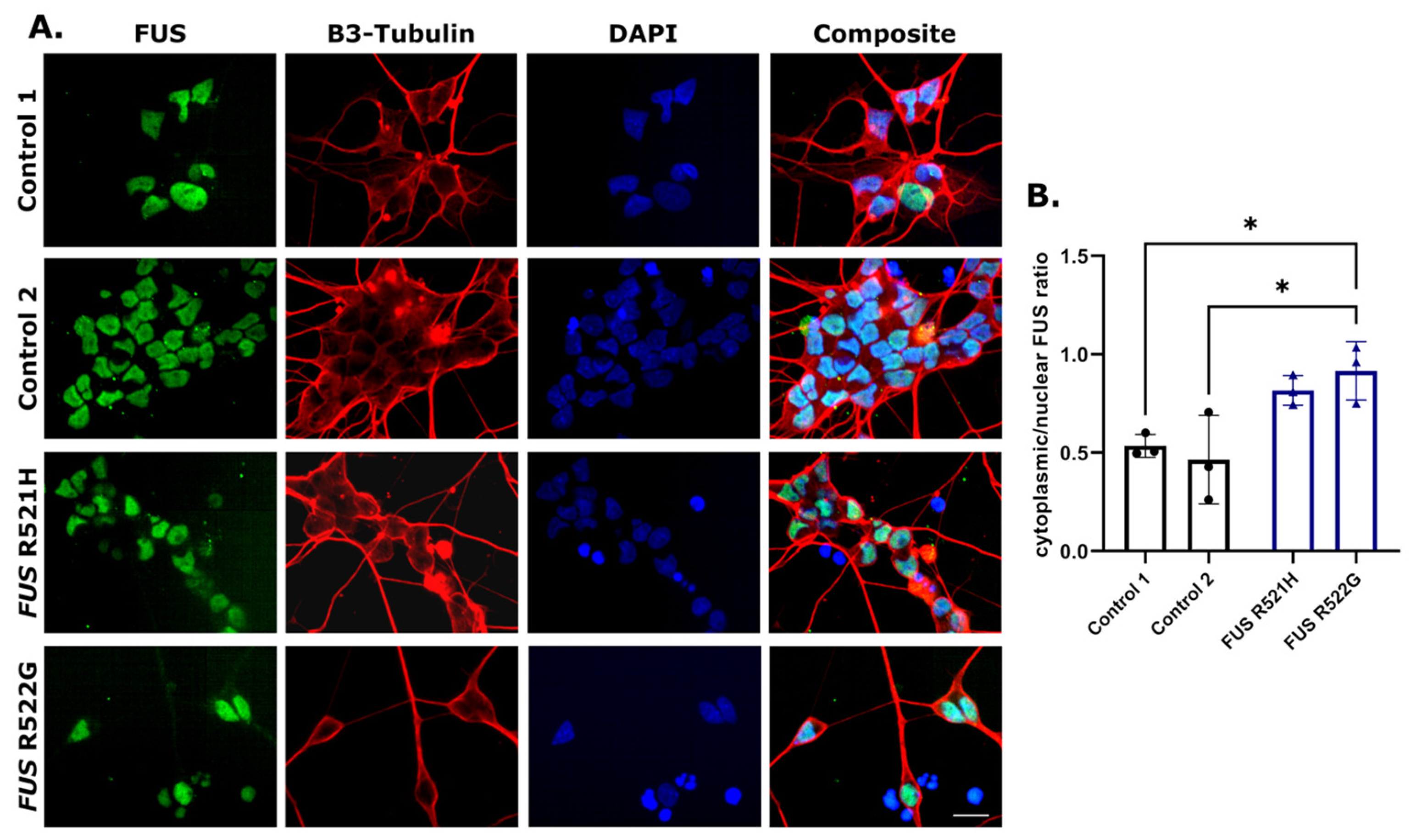
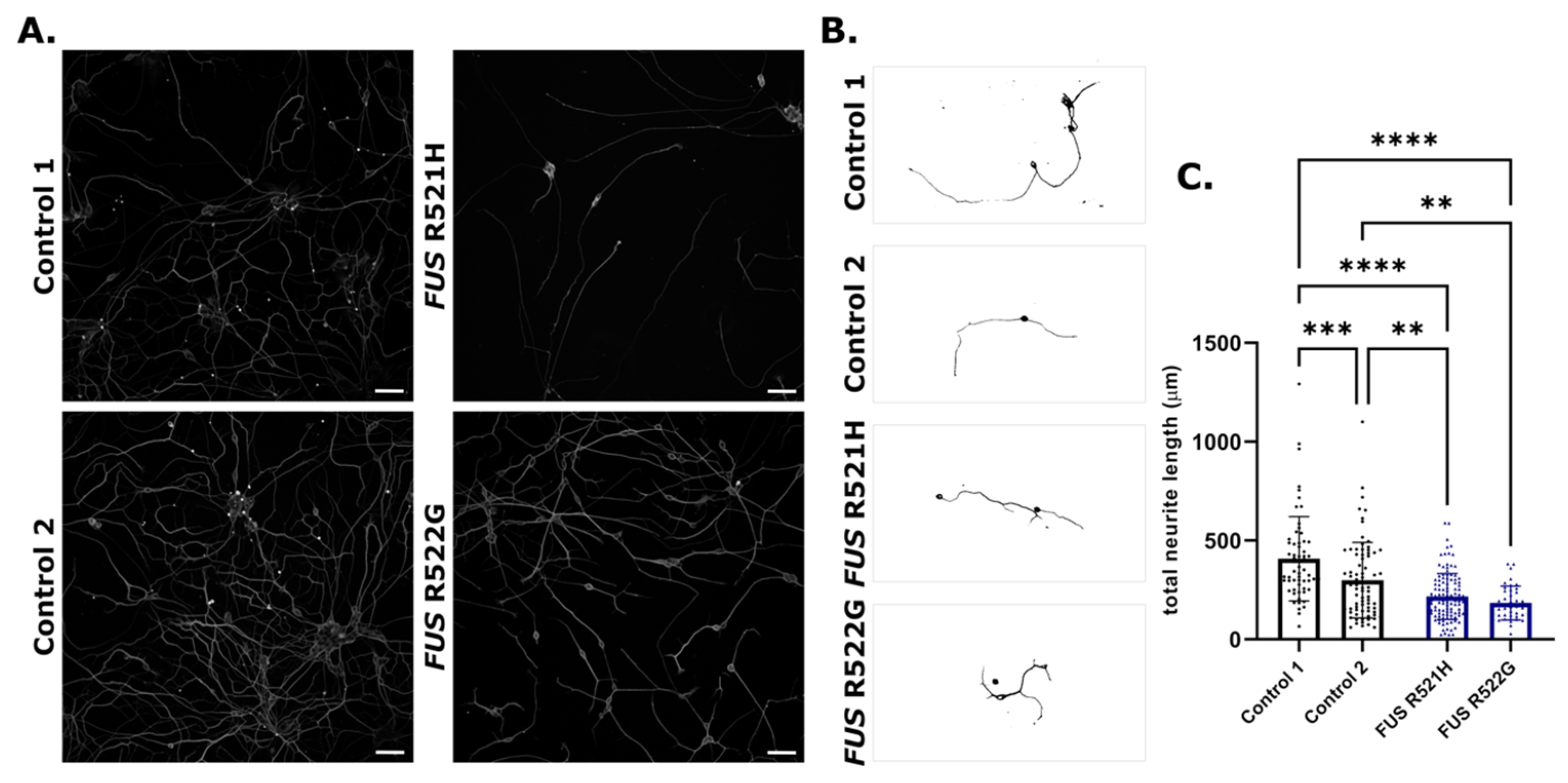
3.3. Phenotypic Screening
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Maury, Y.; Côme, J.; Piskorowski, R.A.; Salah-Mohellibi, N.; Chevaleyre, V.; Peschanski, M.; Martinat, C.; Nedelec, S. Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat. Biotechnol. 2015, 33, 89–96. [Google Scholar] [CrossRef]
- Fernandopulle, M.S.; Prestil, R.; Grunseich, C.; Wang, C.; Gan, L.; Ward, M. Transcription Factor-Mediated Differentiation of Human iPSCs into Neurons. Curr. Protoc. Cell Biol. 2018, 79, e51. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug. Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Penney, J.; Ralvenius, W.T.; Tsai, L.H. Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol. Psychiatry 2020, 25, 148–167. [Google Scholar] [CrossRef]
- Laperle, A.H.; Sances, S.; Yucer, N.; Dardov, V.J.; Garcia, V.J.; Ho, R.; Fulton, A.N.; Jones, M.R.; Roxas, K.M.; Avalos, P.; et al. iPSC modeling of young-onset Parkinson’s disease reveals a molecular signature of disease and novel therapeutic candidates. Nat. Med. 2020, 26, 289–299. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Maragakis, N.J. Mini-Review: Induced pluripotent stem cells and the search for new cell-specific ALS therapeutic targets. Neurosci. Lett. 2021, 755, 135911. [Google Scholar] [CrossRef]
- Ramos, D.M.; Skarnes, W.C.; Singleton, A.B.; Cookson, M.R.; Ward, M.E. Tackling neurodegenerative diseases with genomic engineering: A new stem cell initiative from the NIH. Neuron 2021, 109, 1080–1083. [Google Scholar] [CrossRef]
- Simkin, D.; Papakis, V.; Bustos, B.I.; Ambrosi, C.M.; Ryan, S.J.; Baru, V.; Williams, L.A.; Dempsey, G.T.; McManus, O.B.; Landers, J.E.; et al. Homozygous might be hemizygous: CRISPR/Cas9 editing in iPSCs results in detrimental on-target defects that escape standard quality controls. Stem Cell Rep. 2022, 17, 993–1008. [Google Scholar] [CrossRef]
- Weisheit, I.; Kroeger, J.A.; Malik, R.; Klimmt, J.; Crusius, D.; Dannert, A.; Dichgans, M.; Paquet, D. Detection of Deleterious On-Target Effects after HDR-Mediated CRISPR Editing. Cell Rep. 2020, 31, 107689. [Google Scholar] [CrossRef]
- Rowland, L.P.; Shneider, N.A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New. Treatment? Front. Aging Neurosci. 2017, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Gowland, A.; Opie-Martin, S.; Scott, K.M.; Jones, A.R.; Mehta, P.; Batts, C.J.; Ellis, C.L.; Leigh, P.N.; Shaw, C.E.; Sreedharan, J.; et al. Predicting the future of ALS: The impact of demographic change and potential new treatments on the prevalence of ALS in the United Kingdom 2020–2116. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 264–274. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Vucic, S.; Talbot, K.; McDermott, C.J.; Hardiman, O.; Shefner, J.M.; Al-Chalabi, A.; Huynh, W.; Cudkowicz, M.; Talman, P.; et al. Improving clinical trial outcomes in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2021, 17, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.A.; Jackson, C.E.; Heiman-Patterson, T.D.; Bettica, P.; Brooks, B.R.; Pioro, E.P. Real-world evidence of riluzole effectiveness in treating amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.; Al Khleifat, A.; Meurgey, J.-H.; Jones, A.; Leigh, P.N.; Bensimon, G.; Al-Chalabi, A. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: A retrospective analysis of data from a dose-ranging study. Lancet Neurol. 2018, 17, 416–422. [Google Scholar] [CrossRef]
- Mora, J.S.; Genge, A.; Chio, A.; Estol, C.J.; Chaverri, D.; Hernández, M.; Marín, S.; Mascias, J.; Rodriguez, G.E.; Povedano, M.; et al. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: A randomized clinical trial. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 5–14. [Google Scholar] [CrossRef]
- Abe, K.; Aoki, M.; Tsuji, S.; Itoyama, Y.; Sobue, G.; Togo, M.; Hamada, C.; Tanaka, M.; Akimoto, M.; Nakamura, K.; et al. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Lunetta, C.; Moglia, C.; Lizio, A.; Caponnetto, C.; Dubbioso, R.; Giannini, F.; Matà, S.; Mazzini, L.; Sabatelli, M.; Siciliano, G.; et al. The Italian multicenter experience with edaravone in amyotrophic lateral sclerosis. J. Neurol. 2020, 267, 3258–3267. [Google Scholar] [CrossRef]
- Ganesalingam, J.; Stahl, D.; Wijesekera, L.; Galtrey, C.; Shaw, C.E.; Leigh, P.N.; Al-Chalabi, A. Latent cluster analysis of ALS phenotypes identifies prognostically differing groups. PLoS ONE 2009, 4, e7107. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Hardiman, O.; Kiernan, M.C.; Chiò, A.; Rix-Brooks, B.; van den Berg, L.H. Amyotrophic lateral sclerosis: Moving towards a new classification system. Lancet Neurol. 2016, 15, 1182–1194. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Krainer, A.R.; Cleveland, D.W. Antisense Oligonucleotide Therapies for Neurodegenerative Diseases. Annu. Rev. Neurosci. 2019, 42, 385–406. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.M.; Cudkowicz, M.E.; Genge, A.; Shaw, P.J.; Sobue, G.; Bucelli, R.C.; Chiò, A.; Van Damme, P.; Ludolph, A.C.; Glass, J.D.; et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2022, 387, 1099–1110. [Google Scholar] [CrossRef]
- Boros, B.D.; Schoch, K.M.; Kreple, C.J.; Miller, T.M. Antisense Oligonucleotides for the Study and Treatment of ALS. Neurotherapeutics 2022, 19, 1145–1158. [Google Scholar] [CrossRef]
- Dimos, J.T.; Rodolfa, K.T.; Niakan, K.K.; Weisenthal, L.M.; Mitsumoto, H.; Chung, W.; Croft, G.F.; Saphier, G.; Leibel, R.; Goland, R.; et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 2008, 321, 1218–1221. [Google Scholar] [CrossRef]
- Donnelly, C.J.; Zhang, P.W.; Pham, J.T.; Haeusler, A.R.; Mistry, N.A.; Vidensky, S.; Daley, E.L.; Poth, E.M.; Hoover, B.; Fines, D.M.; et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013, 80, 415–428. [Google Scholar] [CrossRef]
- Bilican, B.; Serio, A.; Barmada, S.J.; Nishimura, A.L.; Sullivan, G.J.; Carrasco, M.; Phatnani, H.P.; Puddifoot, C.A.; Story, D.; Fletcher, J.; et al. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc. Natl. Acad. Sci. USA 2012, 109, 5803–5808. [Google Scholar] [CrossRef]
- Selvaraj, B.T.; Livesey, M.R.; Zhao, C.; Gregory, J.M.; James, O.T.; Cleary, E.M.; Chouhan, A.K.; Gane, A.B.; Perkins, E.M.; Dando, O.; et al. C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca(2+)-permeable AMPA receptor-mediated excitotoxicity. Nat. Commun. 2018, 9, 347. [Google Scholar] [CrossRef]
- Smith, L.; Cupid, B.C.; Dickie, B.G.M.; Al-Chalabi, A.; Morrison, K.E.; Shaw, C.E.; Shaw, P.J. Establishing the UK DNA Bank for motor neuron disease (MND). BMC Genet. 2015, 16, 84. [Google Scholar] [CrossRef]
- Hui-Yuen, J.; McAllister, S.; Koganti, S.; Hill, E.; Bhaduri-McIntosh, S. Establishment of Epstein-Barr virus growth-transformed lymphoblastoid cell lines. J. Vis. Exp. 2011, 57, 3321. [Google Scholar]
- Nam, H.-Y.; Shim, S.-M.; Han, B.-G.; Jeon, J.-P. Human lymphoblastoid cell lines: A goldmine for the biobankomics era. Pharmacogenomics 2011, 12, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Melo, U.S.; Leite, F.D.S.; Costa, S.; Rosenberg, C.; Zatz, M. A fast method to reprogram and CRISPR/Cas9 gene editing from erythroblasts. Stem Cell Res. 2018, 31, 52–54. [Google Scholar] [CrossRef]
- Rajesh, D.; Dickerson, S.J.; Yu, J.; Brown, M.E.; Thomson, J.A.; Seay, N.J. Human lymphoblastoid B-cell lines reprogrammed to EBV-free induced pluripotent stem cells. Blood 2011, 118, 1797–1800. [Google Scholar] [CrossRef]
- Barrett, R.; Ornelas, L.; Yeager, N.; Mandefro, B.; Sahabian, A.; Lenaeus, L.; Targan, S.R.; Svendsen, C.N.; Sareen, D. Reliable generation of induced pluripotent stem cells from human lymphoblastoid cell lines. Stem Cells Transl. Med. 2014, 3, 1429–1434. [Google Scholar] [CrossRef] [PubMed]
- Hedges, E.C.; Topp, S.; Shaw, C.E.; Nishimura, A.L. Generation of six induced pluripotent stem cell lines from patients with amyotrophic lateral sclerosis with associated genetic mutations in either FUS or ANXA11. Stem Cell Res. 2021, 52, 102246. [Google Scholar] [CrossRef]
- Du, Z.-W.; Chen, H.; Liu, H.; Lu, J.; Qian, K.; Huang, C.-L.; Zhong, X.; Fan, F.; Zhang, S.-C. Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nat. Com. 2015, 25, 6626. [Google Scholar] [CrossRef]
- Mrozek-Gorska, P.; Buschle, A.; Pich, D.; Schwarzmayr, T.; Fechtner, R.; Scialdone, A.; Hammerschmidt, W. Epstein-Barr virus reprograms human B lymphocytes immediately in the prelatent phase of infection. Proc. Natl. Acad. Sci. USA 2019, 116, 16046–16055. [Google Scholar] [CrossRef]
- Smatti, M.K.; Al-Sadeq, D.W.; Ali, N.H.; Pintus, G.; Abou-Saleh, H.; Nasrallah, G.K. Epstein–Barr Virus Epidemiology, Serology, and Genetic Variability of LMP-1 Oncogene Among Healthy Population: An Update. Front. Oncol. 2018, 8, 211. [Google Scholar] [CrossRef]
- Jox, A.; Rohen, C.; Belge, G.; Bartnitzke, S.; Pawlita, M.; Diehl, V.; Bullerdiek, J.; Wolf, J. Integration of Epstein-Barr virus in Burkitt’s lymphoma cells leads to a region of enhanced chromosome instability. Ann. Oncol. 1997, 8, 131–135. [Google Scholar] [CrossRef]
- Janjetovic, S.; Hinke, J.; Balachandran, S.; Akyüz, N.; Behrmann, P.; Bokemeyer, C.; Dierlamm, J.; Penas, E.M.M. Non-Random Pattern of Integration for Epstein-Barr Virus with Preference for Gene-Poor Genomic Chromosomal Regions into the Genome of Burkitt Lymphoma Cell Lines. Viruses 2022, 14, 86. [Google Scholar] [CrossRef]
- Xu, M.; Zhang, W.-L.; Zhu, Q.; Yao, Y.-Y.; Feng, Q.-S.; Zhang, Z.; Peng, R.-J.; Jia, W.-H.; He, G.-P.; Feng, L.; et al. Genome-wide profiling of Epstein-Barr virus integration by targeted sequencing in Epstein-Barr virus associated malignancies. Theranostics 2019, 9, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Coppotelli, G.; Mughal, N.; Callegari, S.; Sompallae, R.; Caja, L.; Luijsterburg, M.S.; Dantuma, N.P.; Moustakas, A.; Masucci, M.G. The Epstein–Barr virus nuclear antigen-1 reprograms transcription by mimicry of high mobility group A proteins. Nucleic Acids Res. 2013, 41, 2950–2962. [Google Scholar] [CrossRef]
- Gaspar-Maia, A.; Alajem, A.; Meshorer, E.; Ramalho-Santos, M. Open chromatin in pluripotency and reprogramming. Nat. Rev. Mol. Cell Biol. 2011, 12, 36–47. [Google Scholar] [CrossRef]
- Dittmer, D.P.; Hilscher, C.J.; Gulley, M.L.; Yang, E.V.; Chen, M.; Glaser, R. Multiple pathways for Epstein-Barr virus episome loss from nasopharyngeal carcinoma. Int. J. Cancer 2008, 123, 2105–2112. [Google Scholar] [CrossRef]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.N.; Topp, S.D.; Fallini, C.; Shibata, H.; Chen, H.-J.; Troakes, C.; King, A.; Ticozzi, N.; Kenna, K.P.; Soragia-Gkazi, A.; et al. Mutations in the vesicular trafficking protein annexin A11 are associated with amyotrophic lateral sclerosis. Sci. Trans. Med. 2017, 9, eaad9157. [Google Scholar] [CrossRef] [PubMed]
- Teyssou, E.; Muratet, F.; Amador, M.-D.; Ferrien, M.; Lautrette, G.; Machat, S.; Boillée, S.; Larmonier, T.; Saker, S.; Leguern, E.; et al. Genetic screening of ANXA11 revealed novel mutations linked to Amyotrophic Lateral Sclerosis. Neurobiol. Aging 2021, 99, e11–e102. [Google Scholar] [CrossRef]
- Zhang, K.; Liu, Q.; Liu, K.; Shen, D.; Tai, H.; Shu, S.; Ding, Q.; Fu, H.; Liu, S.; Wang, Z.; et al. ANXA11 mutations prevail in Chinese ALS patients with and without cognitive dementia. Neurol. Genet. 2018, 4, e237. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Moll, T.; Ramesh, T.; Castelli, L.; Beer, A.; Robins, H.; Fox, I.; Niedermoser, I.; Van Damme, P.; Moisse, M.; et al. Mutations in the Glycosyltransferase Domain of GLT8D1 Are Associated with Familial Amyotrophic Lateral Sclerosis. Cell Rep. 2019, 26, 2298–2306.e5. [Google Scholar] [CrossRef]
- Fat, S.C.M.; McCann, E.P.; Williams, K.L.; Henden, L.; Twine, N.A.; Bauer, D.C.; Pamphlett, R.; Kiernan, M.C.; Rowe, D.B.; Nicholson, G.A.; et al. Genetic analysis of GLT8D1 and ARPP21 in Australian familial and sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2021, 101, e9–e297. [Google Scholar]
- Li, W.; Liu, Z.; Sun, W.; Yuan, Y.; Hu, Y.; Ni, J.; Jiao, B.; Fang, L.; Li, J.; Shen, L.; et al. Mutation analysis of GLT8D1 and ARPP21 genes in amyotrophic lateral sclerosis patients from mainland China. Neurobiol. Aging 2020, 85, 156.e1–156.e4. [Google Scholar] [CrossRef]
- Lamas, N.J.; Roybon, L. Harnessing the Potential of Human Pluripotent Stem Cell-Derived Motor Neurons for Drug Discovery in Amyotrophic Lateral Sclerosis: From the Clinic to the Laboratory and Back to the Patient. Front. Drug Discov. 2021, 1, 773424. [Google Scholar] [CrossRef]
- Richard, J.-P.; Maragakis, N.J. Induced pluripotent stem cells from ALS patients for disease modeling. Brain Res. 2015, 1607, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Fumagalli, L.; Prior, R.; Van Den Bosch, L. Current Advances and Limitations in Modeling ALS/FTD in a Dish Using Induced Pluripotent Stem Cells. Front. Neurosci. 2017, 11, 671. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovás, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Com. 2017, 8, 861. [Google Scholar] [CrossRef]
- Liu, M.-L.; Zang, T.; Zhang, C.-L. Direct Lineage Reprogramming Reveals Disease-Specific Phenotypes of Motor Neurons from Human ALS Patients. Cell Rep. 2016, 14, 115–128. [Google Scholar] [CrossRef]
- Liu, X.; Chen, J.; Liu, W.; Li, X.; Chen, Q.; Liu, T.; Gao, S.; Deng, M. The fused in sarcoma protein forms cytoplasmic aggregates in motor neurons derived from integration-free induced pluripotent stem cells generated from a patient with familial amyotrophic lateral sclerosis carrying the FUS-P525L mutation. Neurogen 2015, 16, 223–231. [Google Scholar] [CrossRef]
- Ichiyanagi, N.; Fujimori, K.; Yano, M.; Ishihara-Fujisaki, C.; Sone, T.; Akiyama, T.; Okada, Y.; Akamatsu, W.; Matsumoto, T.; Ishikawa, M.; et al. Establishment of In Vitro FUS-Associated Familial Amyotrophic Lateral Sclerosis Model Using Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2016, 6, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Garone, M.G.; Birsa, N.; Rosito, M.; Salaris, F.; Mochi, M.; de Turris, V.; Nair, R.R.; Cunningham, T.J.; Fisher, E.M.C.; Morlando, M.; et al. ALS-related FUS mutations alter axon growth in motoneurons and affect HuD/ELAVL4 and FMRP activity. Commun. Biol. 2021, 4, 1025. [Google Scholar] [CrossRef]
- Akiyama, T.; Suzuki, N.; Ishikawa, M.; Fujimori, K.; Sone, T.; Kawada, J.; Funayama, R.; Fujishima, F.; Mitsuzawa, S.; Ikeda, K.; et al. Aberrant axon branching via Fos-B dysregulation in FUS-ALS motor neurons. EBioMedicine 2019, 45, 362–378. [Google Scholar] [CrossRef] [PubMed]
- Dittlau, K.S.; Krasnow, E.N.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Kerstens, A.; Giacomazzi, G.; Pavie, B.; Rossaert, E.; Beckers, J.; et al. Human motor units in microfluidic devices are impaired by FUS mutations and improved by HDAC6 inhibition. Stem Cell Rep. 2021, 16, 2213–2227. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MNDA Cell Line ID | Status | Gene | Mutation | Age at Collection | Sex |
|---|---|---|---|---|---|
| LC0501 | Control | − | − | 69 | Female |
| SNC0106 | Control | − | − | 71 | Female |
| BLI0083 | Control | − | − | 72 | Female |
| BC6325 | Control | − | − | 83 | Female |
| SC3709 | Control | − | − | 84 | Female |
| LNH0108 | Control | − | − | 61 | Male |
| LCA0042 | Control | − | − | 61 | Male |
| LC0209 | Control | − | − | 71 | Male |
| BC6055 | Control | − | − | 74 | Male |
| SC3602 | Control | − | − | 64 | Male |
| LP0584 | Familial | ANXA11 | D40G | 75 | Female |
| LP0582 | Familial | ANXA11 | D40G | 76 | Female |
| SMA0020 | Familial | ANXA11 | G38R | 51 | Male |
| LP0663 | Familial | ANXA11 | G38R | 63 | Male |
| LP0039 | Sporadic | ANXA11 | R235Q | 66 | Female |
| BP6184 | Sporadic | ARPP21 | P713L | 59 | Male |
| SP3185 | Sporadic | ARPP21 | P529L | 45 | Male |
| LP0225 | Familial | ARPP21 | P529L | 41 | Female |
| SP3277 | Familial | ARPP21 | P529L | 45 | Male |
| SMA0078 | Familial | TARDBP | N378D | 60 | Male |
| SP3068 | Familial | TARDBP | M337V | 59 | Male |
| SP3154 | Familial | TARDBP | G348V | 58 | Male |
| BOX0029 | Familial | C9ORF72 | G4C2 intronic expansion | 58 | Female |
| LPO0036 | Familial | C9ORF72 | G4C2 intronic expansion | 64 | Male |
| BP6021 | Sporadic | C9ORF72 | G4C2 intronic expansion | 38 | Female |
| BP6204 | Sporadic | C9ORF72 | G4C2 intronic expansion | 45 | Female |
| LP0393 | Familial | unknown | - | 71 | Female |
| LP0048 | Familial | FUS | R522G | 29 | Male |
| LP0051 | Familial | FUS | Q519E | 52 | Male |
| LP0168 | Familial | FUS | R521H | 44 | Female |
| MNDA Cell Line ID | iPSC Derivation Method | Parent Cell Type | Number of Clones Frozen | Characterisation | |||||
|---|---|---|---|---|---|---|---|---|---|
| Pluripotency ICC | Embryoid Body Assay | Karyotyping | EBV Loss (Passage of Negative Stocks) | Confirmation of ALS Mutation | STR Profilling | ||||
| LC0501 | P | LCL | 8 | ✓ | ✓ | G | P30 | n/a | ✓ |
| SNC0106 | P | LCL | 10 | ✓ | ✓ | G | P13 | n/a | ✓ |
| BLI0083 | P | LCL | 5 | ✓ | ✓ | KS * | P16 | n/a | ✓ |
| BC6325 | P | LCL | 3 | ✓ | ✓ | KS | P20 | n/a | ✓ |
| SC3709 | SeV | LCL | 4 | ✓ | ✓ | G | P22 | n/a | ✓ |
| LNH0108 | P | LCL | 12 | ✓ | ✓ | G | P28 | n/a | ✓ |
| LCA0042 | P | LCL | 20 | ✓ | ✓ | G | P30 | n/a | ✓ |
| LC0209 | P | LCL | 7 | ✓ | ✓ | G | P32 | n/a | ✓ |
| BC6055 | P | LCL | 1 | ✓ | ✓ | KS | EBV present | n/a | ✓ |
| SC3602 | P | LCL | 20 | ✓ | ✓ | KS | EBV present | n/a | ✓ |
| LP0584 | P | LCL | 5 | ✓ | ✓ | G | P17 | ✓ | ✓ |
| LP0582 | P | LCL | 5 | ✓ | ✓ | G ** | P23 | ✓ | ✓ |
| SMA0020 | SeV | LCL | 5 | ✓ | ✓ | G | P26 | ✓ | ✓ |
| LP0663 | P | LCL | 3 | ✓ | ✓ | G | P15 | ✓ | ✓ |
| LP0039 | P | LCL | 4 | ✓ | ✓ | G | P26 | ✓ | ✓ |
| BP6184 | P | LCL | 14 | ✓ | ✓ | G | P20 | ✓ | ✓ |
| SP3185 | P | LCL | 12 | ✓ | ✓ | G | EBV present | ✓ | ✓ |
| LP0225 | P | LCL | 10 | ✓ | ✓ | KS | EBV present | ✓ | ✓ |
| SP3277 | P | LCL | 9 | ✓ | ✓ | KS | P10 | ✓ | ✓ |
| SMA0078 | P | LCL | 6 | ✓ | ✓ | KS | P29 | ✓ | ✓ |
| SP3068 | P | LCL | 21 | ✓ | ✓ | G | P19 | ✓ | ✓ |
| SP3154 | P | LCL | 5 | ✓ | ✓ | KS | P12 | ✓ | ✓ |
| BOX0029 | P | LCL | 13 | ✓ | ✓ | G | P24 | ✓ | ✓ |
| LPO0036 | P | LCL | 10 | ✓ | ✓ | KS | P30 | ✓ | ✓ |
| BP6021 | P | LCL | 6 | ✓ | ✓ | G | P30 | ✓ | ✓ |
| BP6204 | P | LCL | 5 | ✓ | ✓ | KS | P19 | ✓ | ✓ |
| LP0393 | P | LCL | 4 | ✓ | ✓ | KS *** | P25 | ✓ | ✓ |
| LP0048 | P | LCL | 3 | ✓ | ✓ | G | P20 | ✓ | ✓ |
| LP0051 | P | LCL | 3 | ✓ | ✓ | G | P19 | ✓ | ✓ |
| LP0168 | P | LCL | 3 | ✓ | ✓ | G | P14 | ✓ | ✓ |
| LCA0042 | P | PBL | 11 | ✓ | ✓ | KS **** | n/a | n/a | ✓ |
| LNH0108 | P | PBL | 10 | ✓ | ✓ | G | n/a | n/a | ✓ |
| SMA0078 | P | PBL | 10 | ✓ | ✓ | KS ***** | n/a | ✓ | ✓ |
| SP3277 | P | PBL | 11 | ✓ | ✓ | G | n/a | ✓ | ✓ |
| SP3154 | P | PBL | 8 | ✓ | ✓ | KS | n/a | ✓ | ✓ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hedges, E.C.; Cocks, G.; Shaw, C.E.; Nishimura, A.L. Generation of an Open-Access Patient-Derived iPSC Biobank for Amyotrophic Lateral Sclerosis Disease Modelling. Genes 2023, 14, 1108. https://doi.org/10.3390/genes14051108
Hedges EC, Cocks G, Shaw CE, Nishimura AL. Generation of an Open-Access Patient-Derived iPSC Biobank for Amyotrophic Lateral Sclerosis Disease Modelling. Genes. 2023; 14(5):1108. https://doi.org/10.3390/genes14051108
Chicago/Turabian StyleHedges, Erin C., Graham Cocks, Christopher E. Shaw, and Agnes L. Nishimura. 2023. "Generation of an Open-Access Patient-Derived iPSC Biobank for Amyotrophic Lateral Sclerosis Disease Modelling" Genes 14, no. 5: 1108. https://doi.org/10.3390/genes14051108