
Germline JAK2 E846D Substitution as the Cause of Erythrocytosis?

 , ,
, ,
Abstract
:1. Introduction
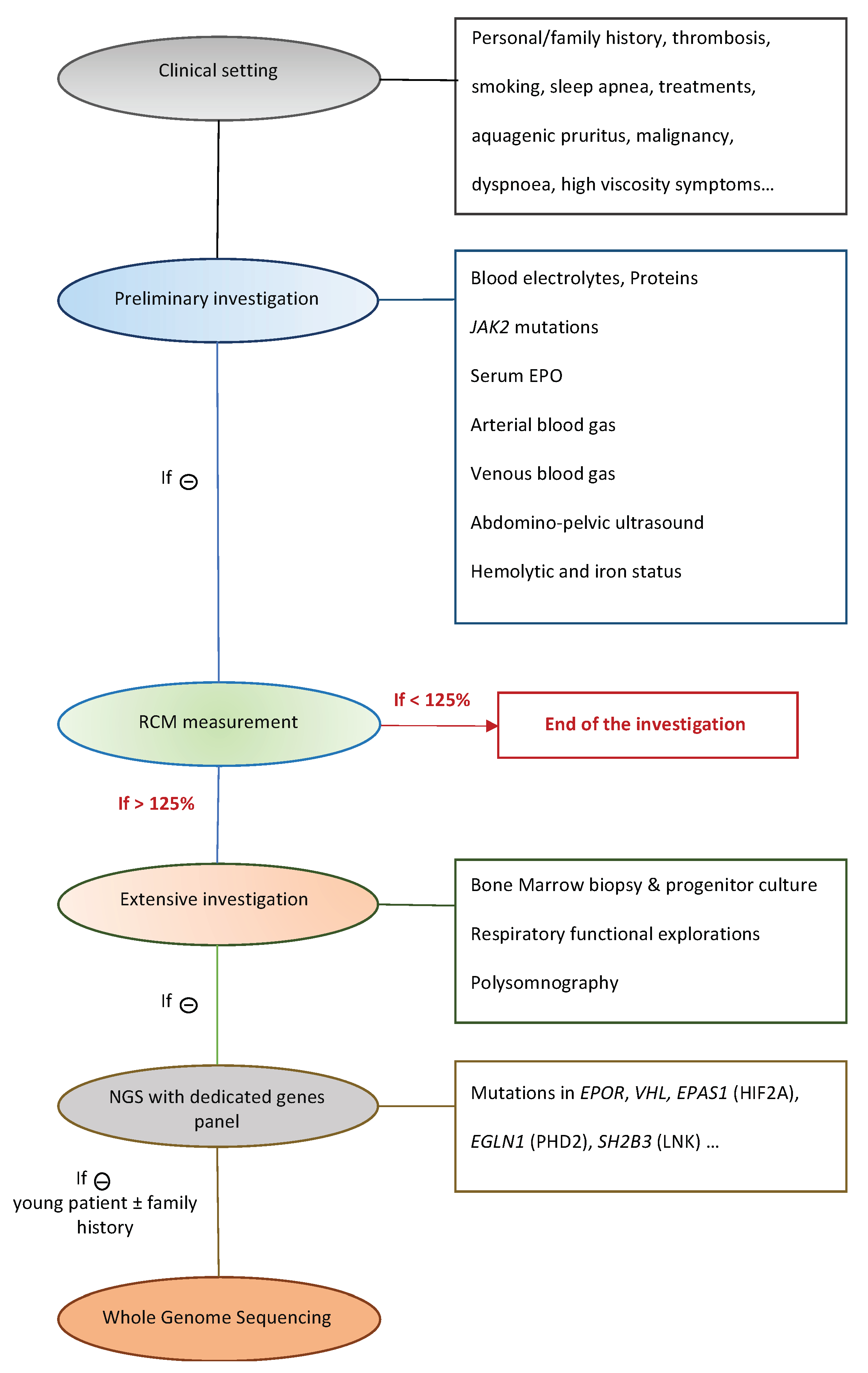
2. Materials and Methods
2.1. Patients
2.2. In Silico Analysis
2.3. Next Generation Sequencing
2.4. Sanger Sequencing for Familial Segregation Analysis
3. Results
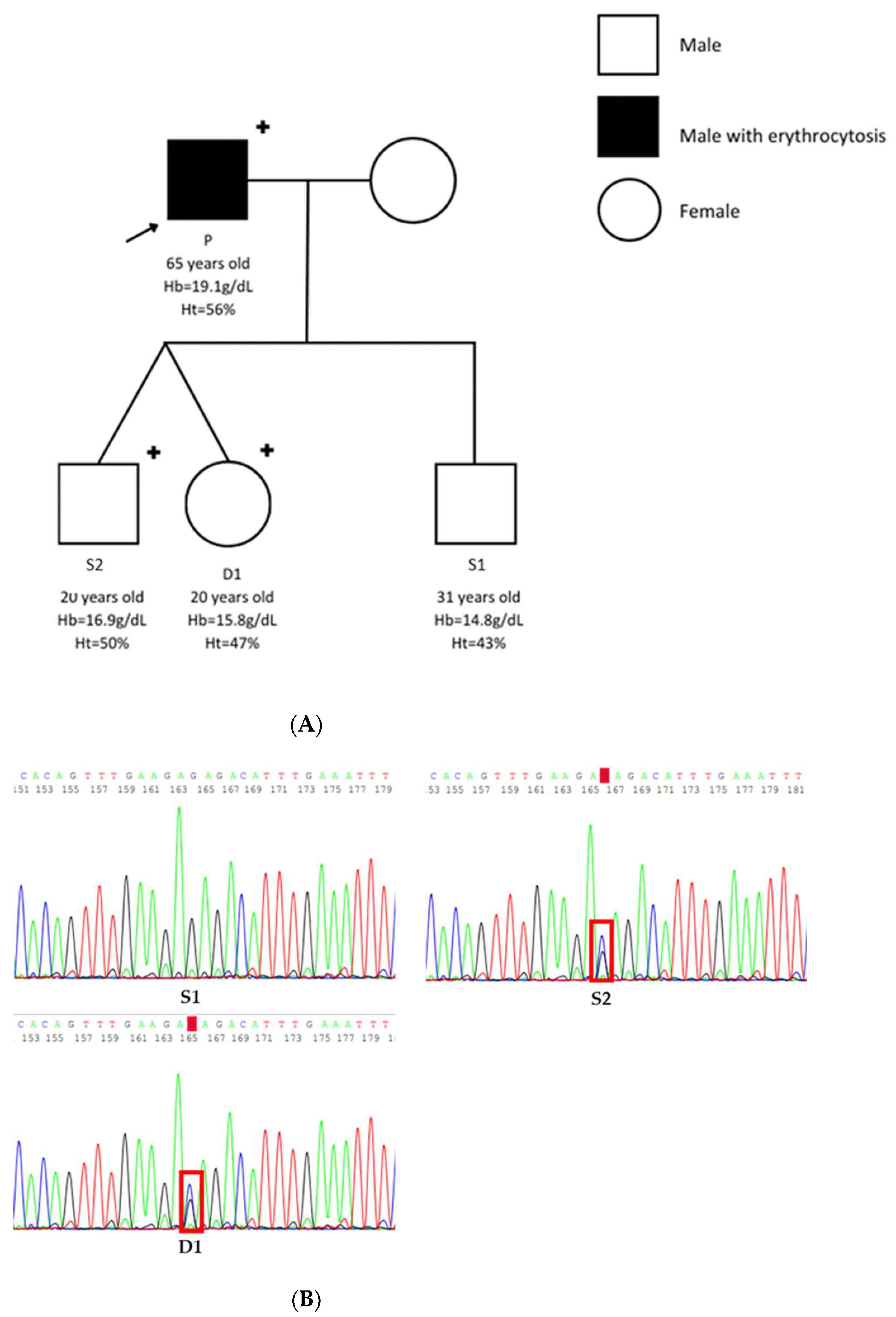
3.1. Patients
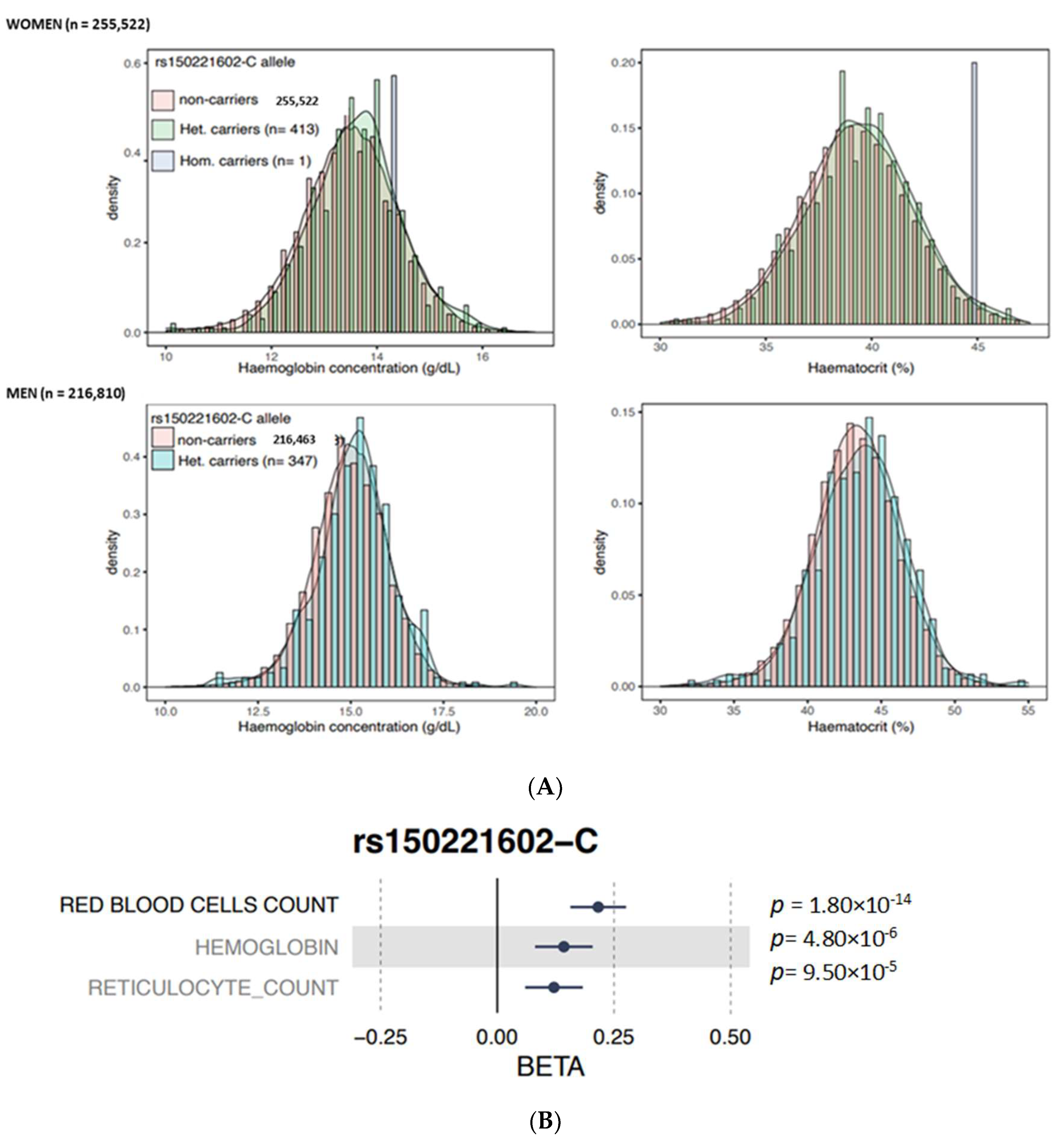
3.2. In Silico Analysis
4. Discussion
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Johansson, P.L. An elevated venous haemoglobin concentration cannot be used as a surrogate marker for absolute erythrocytosis: A study of patients with polycythaemia vera and apparent polycythaemia. Br. J. Haematol. 2005, 129, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Thiele, J.; Vannucchi, A.M.; Tefferi, A. Rethinking the diagnostic criteria of polycythemia vera. Leukemia 2013, 28, 1191–1195. [Google Scholar] [CrossRef] [PubMed]
- Bader, M.S.; Meyer, S.C. JAK2 in Myeloproliferative Neoplasms: Still a Protagonist. Pharmaceuticals 2022, 15, 160. [Google Scholar] [CrossRef]
- Skoda, R.C.; Duek, A.; Grisouard, J. Pathogenesis of myeloproliferative neoplasms. Exp. Hematol. 2015, 43, 599–608. [Google Scholar] [CrossRef] [Green Version]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak–STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [Green Version]
- Varghese, L.N.; Ungureanu, D.; Liau, N.P.D.; Young, S.N.; Laktyushin, A.; Hammaren, H.; Lucet, I.S.; Nicola, N.A.; Silvennoinen, O.; Babon, J.J.; et al. Mechanistic insights into activation and SOCS3-mediated inhibition of myeloproliferative neoplasm-associated JAK2 mutants from biochemical and structural analyses. Biochem. J. 2014, 458, 395–405. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Qu, C.K. Protein tyrosine phosphatases in the JAK/STAT pathway. Front. Biosci. 2008, 13, 4925–4932. [Google Scholar] [CrossRef] [Green Version]
- Maslah, N.; Cassinat, B.; Verger, E.; Kiladjian, J.J.; Velazquez, L. The role of LNK/SH2B3 genetic alterations in myeloproliferative neoplasms and other hematological disorders. Leukemia 2017, 31, 1661–1670. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- James, C.; Ugo, V.; Le Couédic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- McMullin, M.F. Diagnostic workflow for hereditary erythrocytosis and thrombocytosis. Hematology 2019, 2019, 391–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marty, C.; Saint-Martin, C.; Pecquet, C.; Grosjean, S.; Saliba, J.; Mouton, C.; Leroy, E.; Harutyunyan, A.S.; Abgrall, J.-F.; Favier, R.; et al. Germ-line JAK2 mutations in the kinase domain are responsible for hereditary thrombocytosis and are resistant to JAK2 and HSP90 inhibitors. Blood 2014, 123, 1372–1383. [Google Scholar] [CrossRef] [Green Version]
- Etheridge, S.L.; Cosgrove, M.E.; Sangkhae, V.; Corbo, L.M.; Roh, M.E.; Seeliger, M.; Chan, E.L.; Hitchcock, I. A novel activating, germline JAK2 mutation, JAK2R564Q, causes familial essential thrombocytosis. Blood 2014, 123, 1059–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Swierczek, S.; Piterkova, L.; Hickman, K.; Wheeler, D.A.; Prchal, J.T. Whole Exome Sequencing of Polycythemia Vera Reveals Novel Recurrent Somatic and Germline Variation. Blood 2012, 120, 705. [Google Scholar] [CrossRef]
- McMullin, M.F.F.; Mead, A.J.; Ali, S.; Cargo, C.; Chen, F.; Ewing, J.; Garg, M.; Godfrey, A.; Knapper, S.; McLornan, D.P.; et al. A guideline for the management of specific situations in polycythaemia vera and secondary erythrocytosis: A British Society for Haematology Guideline. Br. J. Haematol. 2019, 184, 161–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangat, N.; Szuber, N.; Tefferi, A. JAK2 unmutated erythrocytosis: 2023 update on diagnosis and management. Am. J. Hematol. 2023. [Google Scholar] [CrossRef]
- Tun, P.W.W.; Buka, R.J.; Graham, J.; Dyer, P. Heterozygous, germline JAK2 E846D substitution as the cause of familial erythrocytosis. Br. J. Haematol. 2022, 198, 923–926. [Google Scholar] [CrossRef]
- Kapralova, K.; Horvathova, M.; Pecquet, C.; Kucerova, J.F.; Pospisilova, D.; Leroy, E.; Kralova, B.; Feenstra, J.D.M.; Schischlik, F.; Kralovics, R.; et al. Cooperation of germ line JAK2 mutations E846D and R1063H in hereditary erythrocytosis with megakaryocytic atypia. Blood 2016, 128, 1418–1423. [Google Scholar] [CrossRef] [Green Version]
- Panovska-Stavridis, I.; Eftimov, A.; Ivanovski, M.; Pivkova-Veljanovska, A.; Cevreska, L.; Hermouet, S.; Dimovski, A.J.; Cevrevska, L. Essential Thrombocythemia Associated With Germline JAK2 G571S Variant and Somatic CALR Type 1 Mutation. Clin. Lymphoma Myeloma Leuk. 2016, 16, e55–e57. [Google Scholar] [CrossRef]
- Filser, M.; Aral, B.; Airaud, F.; Chauveau, A.; Bruce, A.; Polfrit, Y.; Thiebaut, A.; Gauthier, M.; Maréchal, C.L.; Lippert, E.; et al. Low incidence of EPOR mutations in idiopathic erythrocytosis. Haematologica 2020, 106, 299–301. [Google Scholar] [CrossRef] [Green Version]
- Gallet, M.; Drouet, C.; Girodon, F.; Nicolas, A.; Riedinger, J.-M. Effect of 99mTc elution in vivo from red cells on red cell volumes measured using autologous 99mTc-labeled red cells: Comparison with 51Cr method. Ann. Biol. Clin. 2020, 78, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Barton, A.R.; Sherman, M.A.; Mukamel, R.E.; Loh, P.-R. Whole-exome imputation within UK Biobank powers rare coding variant association and fine-mapping analyses. Nat. Genet. 2021, 53, 1260–1269. [Google Scholar] [CrossRef]
- Backman, J.D.; Li, A.H.; Marcketta, A.; Sun, D.; Mbatchou, J.; Kessler, M.D.; Benner, C.; Liu, D.; Locke, A.E.; Balasubramanian, S.; et al. Exome sequencing and analysis of 454,787 UK Biobank participants. Nature 2021, 599, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Van Hout, C.V.; Tachmazidou, I.; Backman, J.D.; Hoffman, J.D.; Liu, D.; Pandey, A.K.; Gonzaga-Jauregui, C.; Khalid, S.; Ye, B.; Banerjee, N.; et al. Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature 2020, 586, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Filser, M.; Gardie, B.; Wemeau, M.; Aguilar-Martinez, P.; Giansily-Blaizot, M.; Girodon, F. Importance of Sequencing HBA1, HBA2 and HBB Genes to Confirm the Diagnosis of High Oxygen Affinity Hemoglobin. Genes 2022, 13, 132. [Google Scholar] [CrossRef]
- Mossuz, P.; Girodon, F.; Donnard, M.; Latger-Cannard, V.; Dobo, I.; Boiret, N.; Lecron, J.C.; Binquet, C.; Barro, C.; Hermouet, S.; et al. Diagnostic value of serum erythropoietin level in patients with absolute erythrocytosis. Haematologica 2004, 89, 1194–1198. [Google Scholar]
- Cleyrat, C.; Jelinek, J.; Girodon, F.; Boissinot, M.; Ponge, T.; Harousseau, J.L.; Issa, J.P.; Hermouet, S. JAK2 mutation and disease phenotype: A double L611V/V617F in cis mutation of JAK2 is associated with isolated erythrocytosis and increased activation of AKT and ERK1/2 rather than STAT5. Leukemia 2010, 24, 1069–1073. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.-H.; Park, T.S.; Maeng, H.-Y.; Sun, Y.-K.; Kim, Y.-A.; Kie, J.-H.; Cho, E.H.; Song, J.; Lee, K.-A.; Suh, B.; et al. JAK2 V617F/C618R mutation in a patient with polycythemia vera:A case study and review of the literature. Cancer Genet. Cytogenet. 2009, 189, 43–47. [Google Scholar] [CrossRef]
- Passamonti, F.; Elena, C.; Schnittger, S.; Skoda, R.C.; Green, A.R.; Girodon, F.; Kiladjian, J.-J.; McMullin, M.F.; Ruggeri, M.; Besses, C.; et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood 2011, 117, 2813–2816. [Google Scholar] [CrossRef] [Green Version]
- Anelli, L.; Orsini, P.; Zagaria, A.; Minervini, A.; Coccaro, N.; Parciante, E.; Minervini, C.F.; Cumbo, C.; Tota, G.; Impera, L.; et al. Erythrocytosis with JAK2 GGCC_46/1 haplotype and without JAK2 V617F mutation is associated with CALR rs1049481_G allele. Leukemia 2021, 35, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Zagaria, A.; Tarantini, F.; Orsini, P.; Anelli, L.; Cumbo, C.; Coccaro, N.; Tota, G.; Minervini, C.F.; Parciante, E.; Conserva, M.R.; et al. The genomic analysis brings a new piece to the molecular jigsaw of idiopathic erythrocytosis. Exp. Hematol. Oncol. 2022, 11, 47. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patient P | Son S1 | Son S2 | Daughter D1 | Reference Range | |
|---|---|---|---|---|---|
| Age (years) | 65 | 31 | 20 | 20 | n/a |
| Sex | Male | Male | Male | Female | n/a |
| Hemoglobin (g/dL) | 19.1 | 14.8 | 16.9 | 15.8 | 11.5–16.0 |
| Hematocrit (%) | 56 | 43 | 50 | 47 | 37.0–47.0 |
| Serum EPO (UI/L) | 6.7 | Not tested | Not tested | Not tested | 2.6–9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maaziz, N.; Garrec, C.; Airaud, F.; Bobée, V.; Contentin, N.; Cayssials, E.; Rimbert, A.; Aral, B.; Bézieau, S.; Gardie, B.; et al. Germline JAK2 E846D Substitution as the Cause of Erythrocytosis? Genes 2023, 14, 1066. https://doi.org/10.3390/genes14051066
Maaziz N, Garrec C, Airaud F, Bobée V, Contentin N, Cayssials E, Rimbert A, Aral B, Bézieau S, Gardie B, et al. Germline JAK2 E846D Substitution as the Cause of Erythrocytosis? Genes. 2023; 14(5):1066. https://doi.org/10.3390/genes14051066
Chicago/Turabian StyleMaaziz, Nada, Céline Garrec, Fabrice Airaud, Victor Bobée, Nathalie Contentin, Emilie Cayssials, Antoine Rimbert, Bernard Aral, Stéphane Bézieau, Betty Gardie, and et al. 2023. "Germline JAK2 E846D Substitution as the Cause of Erythrocytosis?" Genes 14, no. 5: 1066. https://doi.org/10.3390/genes14051066