

The Application of Optical Genome Mapping (OGM) in Severe Short Stature Caused by Duplication of 15q14q21.3
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Whole-Exome Sequencing (MGISEQ-2000)
2.3. Copy Number Variation Sequencing (CNV-Seq)
2.4. Optical Genome Mapping (OGM)
2.5. Long-Range PCR and Sanger Sequencing
2.6. Literature Review and Database Search
3. Results
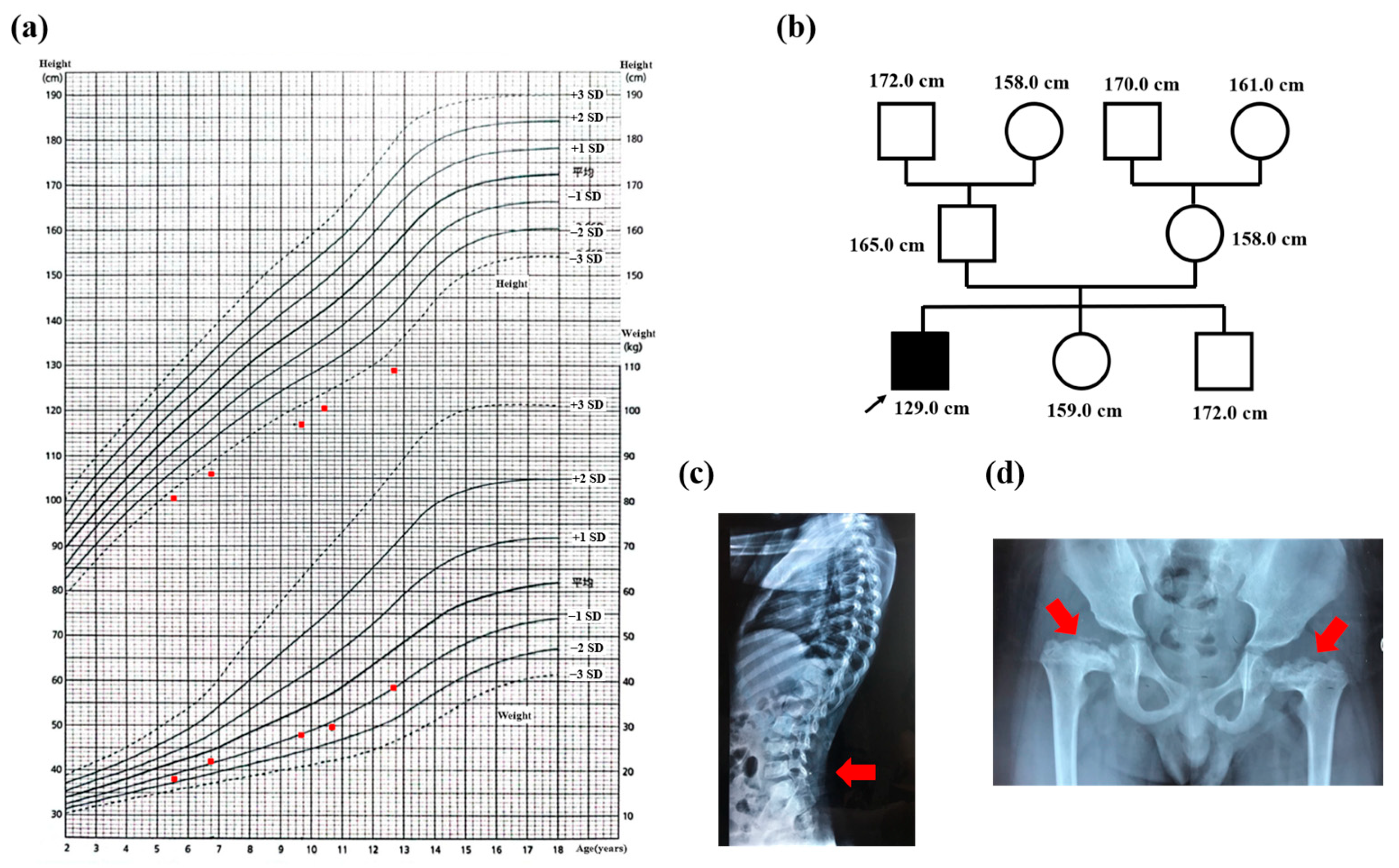
3.1. Case Presentation
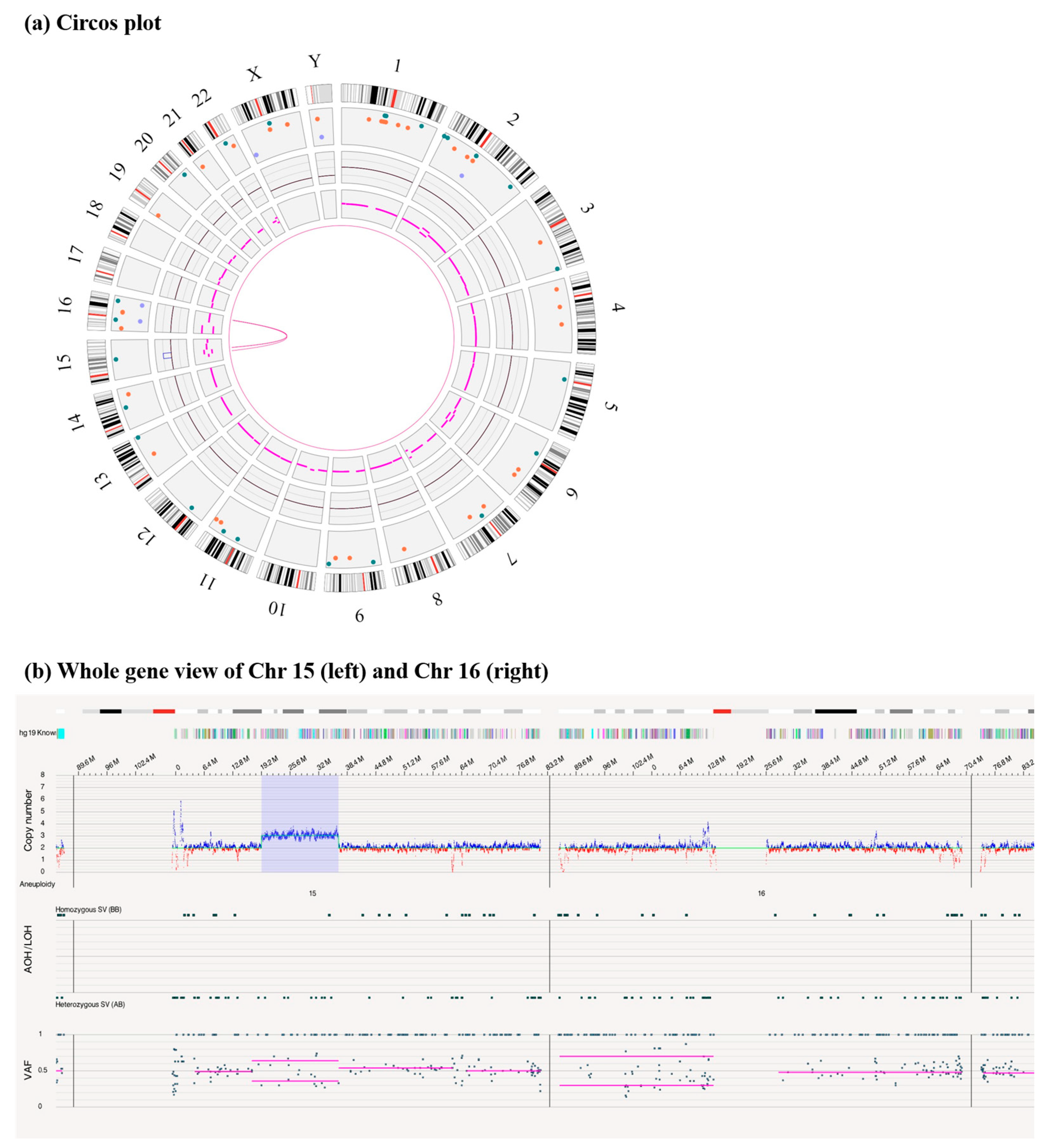
3.2. A 17.27 Mb Duplication of the Long Arm of Chromosome 15 and an Insertion at the Long Arm of Chromosome 16
3.3. A Duplication of 15q14q21.3 Inversely Inserted to 16q23.1 by OGM
3.4. Clinical Features of Patients with Duplication within the 15q14q21.3 Region
4. Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Gender | Age | Bands (Location) | Length | Origin | Methods | Phenotypes |
|---|---|---|---|---|---|---|---|
| Patient 1 [32] | boy | 12 y | 15q14.1q21.1 (NA) | 15 Mb | De novo | High-resolution G-banded cytogenetic analysis | Distinctive face including a narrow forehead, high nasal bridge, narrow palate, dental malocclusion, small ears, and micrognathia; seizure; short stature; cryptorchidism, scrotal hypoplasia, and micro-penis; mental and developmental retardation; others: lumbar kyphosis, bell-shaped chest, mild scoliosis |
| Patient 2 [33] | boy | 14 y | 15q15.3q21.2 (chr15: 44,143,547–50,572,601) | 6.4 Mb | De novo | Chromosomal microarray analysis | Distinctive facial features including macrocephaly, broad forehead, deep-set and widely spaced eyes, broad nose bridge, shallow philtrum, and thick lips; severe short stature; delayed bone age; endocrine: hypogonadism, micro-penis, small testes; global developmental delay and intellectual disability |
| Patient 3 [34] | boy | 8 y | 15q21.2 (chr15: 50,382,769–51,568,204) | 1.1 Mb | De novo | Single-nucleotide polymorphism microarray | Gynecomastia; tall stature and bone age advancement |
| our patient | boy | 10.7 y | 15q14q21.3 (chr15: 40,044,538–57,297,589) | 17.2 Mb | De novo | Karyotype, WES, CNV-seq, and OGM | Severe short stature; distinctive face: a short and flat nose; mild to moderate tricuspid regurgitation; lumbar lordosis and epiphyseal dysplasia of bilateral femur; growth hormone deficiency |
| nssv 3395697 | - | - | 15q21.1 (chr15: 48,728,235–48,793,803) | 65.6 Kb | NA | Clinical Cytogenomic Testing (Postnatal) | Developmental delay AND/OR other significant developmental or morphological phenotypes |
| nssv 575513 | - | - | 15q21.1 (chr15: 48,728,235–48,793,803) | 65.6 Kb | NA | Clinical Cytogenomic Testing (Postnatal) | Global developmental delay |
| nssv 578686 | - | - | 15q14q15.1 (chr15: 36,824,194–41,079,736) | 4.256 Mb | NA | Clinical Cytogenomic Testing (Postnatal) | Developmental delay AND/OR other significant developmental or morphological phenotypes |
| nssv 578687 | - | - | 15q21.3 (chr15: 55,571,230–55,914,205) | 0.343 Mb | NA | NA | Abnormal facial shape, Abnormal heart morphology, Developmental delay AND/OR other significant developmental or morphological phenotypes, Gastroschisis, Global developmental delay |
| Decipher 342055 | - | - | 15q21.3 (chr15: 54,534,868–55,384,248) | 0.849 Mb | De novo | Microarray | Global developmental delay |
| Decipher 331008 | - | - | 15q15.1q15.2 (chr15: 42,621,710–43,056,143) | 0.434 Mb | Maternally inherited | Microarray | Abnormal facial shape, tetralogy of Fallot |
| Decipher 308278 | - | - | 15q21.1 (chr15: 47,384,497–48,918,698) | 1.534 Mb | NA | Microarray | Disproportionate tall stature |
| Decipher 401711 | - | - | 15q15.2q21.2 (chr15: 43,325,133–51,471,260) | 8.146 Mb | De novo | Microarray | Delayed skeletal maturation, hypertelorism, intellectual disability, large earlobe, Macrotia, Pes planus, Proportionate short stature, Proptosis, Thick eyebrow, up-slanted palpebral fissure, Wide mouth |
| Decipher 303564 | - | - | 15q15.3q21.1 (chr15: 44,792,878–45,568,844) | 0.776 Mb | NA | Microarray | Cognitive impairment |
| Decipher 385178 | - | - | 15q15.3q21.1 (chr15: 43,851,578–48,145,280) | 0.385 Mb | NA | Microarray | Delayed ability to walk, short stature |
| Phenotypes | N/Total (%) |
|---|---|
| Neurologic | 10/14 (71.4) |
| Growth | 7/14 (50.0) |
| Short stature | 5/7 (71.4) |
| Tall stature | 2/7 (28.6) |
| Facial abnormalities | 6/14 (42.9) |
| Skeletal abnormalities | 5/14 (35.7) |
| Genitourinary | 3/14 (21.4) |
| Cardiovascular | 3/14 (21.4) |
| Endocrine disorders | 2/14 (14.3) |
| Abdomen | 1/14 (7.1) |
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Collett-Solberg, P.F.; Ambler, G.; Backeljauw, P.F.; Bidlingmaier, M.; Biller, B.M.; Boguszewski, M.C.; Cheung, P.T.; Choong, C.S.Y.; Cohen, L.E.; Cohen, P.; et al. Diagnosis, Genetics, and Therapy of Short Stature in Children: A Growth Hormone Research Society International Perspective. Horm. Res. Paediatr. 2019, 92, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.G.; Clayton, P.E.; Chernausek, S.D. A genetic approach to evaluation of short stature of undetermined cause. Lancet Diabetes Endocrinol. 2018, 6, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Kärkinen, J.; Miettinen, P.J.; Raivio, T.; Hero, M. Etiology of severe short stature below −3 SDS in a screened Finnish population. Eur. J. Endocrinol. 2020, 183, 481–488. [Google Scholar] [CrossRef]
- Jee, Y.H.; Baron, J.; Nilsson, O. New developments in the genetic diagnosis of short stature. Curr. Opin. Pediatr. 2018, 30, 541–547. [Google Scholar] [CrossRef]
- Tyson, C.; Harvard, C.; Locker, R.; Friedman, J.; Langlois, S.; Lewis, M.; Van Allen, M.; Somerville, M.; Arbour, L.; Clarke, L.; et al. Submicroscopic deletions and duplications in individuals with intellectual disability detected by array-CGH. Am. J. Med. Genet. Part A 2005, 139A, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Khetarpal, P.; Das, S.; Panigrahi, I.; Munshi, A. Primordial dwarfism: Overview of clinical and genetic aspects. Mol. Genet. Genom. 2015, 291, 1–15. [Google Scholar] [CrossRef]
- Dauber, A.; Yu, Y.; Turchin, M.C.; Chiang, C.W.; Meng, Y.A.; Demerath, E.W.; Patel, S.R.; Rich, S.S.; Rotter, J.I.; Schreiner, P.J.; et al. Genome-wide Association of Copy-Number Variation Reveals an Association between Short Stature and the Presence of Low-Frequency Genomic Deletions. Am. J. Hum. Genet. 2011, 89, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Wakeling, E.L.; Brioude, F.; Lokulo-Sodipe, O.; O’Connell, S.M.; Salem, J.; Bliek, J.; Canton, A.P.M.; Chrzanowska, K.H.; Davies, J.H.; Dias, R.P.; et al. Diagnosis and management of Silver–Russell syndrome: First international consensus statement. Nat. Rev. Endocrinol. 2017, 13, 105–124. [Google Scholar] [CrossRef] [PubMed]
- Gravholt, C.H.; Viuff, M.H.; Brun, S.; Stochholm, K.; Andersen, N.H. Turner syndrome: Mechanisms and management. Nat. Rev. Endocrinol. 2019, 15, 601–614. [Google Scholar] [CrossRef]
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef]
- Neveling, K.; Mantere, T.; Vermeulen, S.; Oorsprong, M.; van Beek, R.; Kater-Baats, E.; Pauper, M.; van der Zande, G.; Smeets, D.; Weghuis, D.O.; et al. Next-generation cytogenetics: Comprehensive assessment of 52 hematological malignancy genomes by optical genome mapping. Am. J. Hum. Genet. 2021, 108, 1423–1435. [Google Scholar] [CrossRef] [PubMed]
- Dremsek, P.; Schwarz, T.; Weil, B.; Malashka, A.; Laccone, F.; Neesen, J. Optical Genome Mapping in Routine Human Genetic Diagnostics—Its Advantages and Limitations. Genes 2021, 12, 1958. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Ju, X.; Yi, X.; Zhu, Q.; Qu, N.; Liu, T.; Chen, Y.; Jiang, H.; Yang, G.; Zhen, R.; et al. Identification of Sequence Variants in Genetic Disease-Causing Genes Using Targeted Next-Generation Sequencing. PLoS ONE 2011, 6, e29500. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef]
- Ghorbani, F.; de Boer-Bergsma, J.; Verschuuren-Bemelmans, C.C.; Pennings, M.; de Boer, E.N.; Kremer, B.; Vanhoutte, E.K.; de Vries, J.J.; van de Berg, R.; Kamsteeg, E.-J.; et al. Prevalence of intronic repeat expansions in RFC1 in Dutch patients with CANVAS and adult-onset ataxia. J. Neurol. 2022, 269, 6086–6093. [Google Scholar] [CrossRef]
- Schnause, A.C.; Komlosi, K.; Herr, B.; Neesen, J.; Dremsek, P.; Schwarz, T.; Tzschach, A.; Jägle, S.; Lausch, E.; Fischer, J.; et al. Marfan Syndrome Caused by Disruption of the FBN1 Gene due to A Reciprocal Chromosome Translocation. Genes 2021, 12, 1836. [Google Scholar] [CrossRef]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef]
- Kaminsky, E.B.; Kaul, V.; Paschall, J.; Church, D.M.; Bunke, B.; Kunig, D.; Moreno-De-Luca, D.; Moreno-De-Luca, A.; Mulle, J.G.; Warren, S.T.; et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Anesthesia Analg. 2011, 13, 777–784. [Google Scholar] [CrossRef]
- Zhou, E.; Hauser, B.R.; Jee, Y.H. Genetic evaluation in children with short stature. Curr. Opin. Pediatr. 2021, 33, 458–463. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; Mcallister, T.M.; Babovic-Vuksanovic, D.; Beck, S.A.; Borad, M.J.; Bryce, A.H.; Chanan-Khan, A.A.; Ferber, M.J.; Fonseca, R.; Johnson, K.J.; et al. Implementing individualized medicine into the medical practice. Am. J. Med Genet. C Semin. Med. Genet. 2014, 166, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Shen, L.; Bootwalla, M.; Quindipan, C.; Tatarinova, T.; Maglinte, D.T.; Buckley, J.; Raca, G.; Saitta, S.C.; Biegel, J.A.; et al. A semiautomated whole-exome sequencing workflow leads to increased diagnostic yield and identification of novel candidate variants. Cold Spring Harb. Mol. Case Stud. 2019, 5, a003756. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, S.; Momozawa, Y.; Liu, X.; Terao, C.; Kubo, M.; Kamatani, Y. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 2019, 20, 117. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Wang, H.; Chen, H.; Jiang, H.; Yuan, J.; Yang, Z.; Wang, W.-J.; Xu, F.; Guo, X.; Cao, Y.; et al. Identification of balanced chromosomal rearrangements previously unknown among participants in the 1000 Genomes Project: Implications for interpretation of structural variation in genomes and the future of clinical cytogenetics. Genet. Med. 2018, 20, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Mantere, T.; Neveling, K.; Pebrel-Richard, C.; Benoist, M.; van der Zande, G.; Kater-Baats, E.; Baatout, I.; van Beek, R.; Yammine, T.; Oorsprong, M.; et al. Optical genome mapping enables constitutional chromosomal aberration detection. Am. J. Hum. Genet. 2021, 108, 1409–1422. [Google Scholar] [CrossRef]
- Barseghyan, H.; Tang, W.; Wang, R.T.; Almalvez, M.; Segura, E.; Bramble, M.S.; Lipson, A.; Douine, E.D.; Lee, H.; Délot, E.C.; et al. Next-generation mapping: A novel approach for detection of pathogenic structural variants with a potential utility in clinical diagnosis. Genome Med. 2017, 9, 90. [Google Scholar] [CrossRef]
- Pastor, S.; Tran, O.; Jin, A.; Carrado, D.; Silva, B.A.; Uppuluri, L.; Abid, H.Z.; Young, E.; Crowley, T.B.; Bailey, A.G.; et al. Optical mapping of the 22q11.2DS region reveals complex repeat structures and preferred locations for non-allelic homologous recombination (NAHR). Sci. Rep. 2020, 10, 12235. [Google Scholar] [CrossRef]
- Roggenbuck, J.A.; Mendelsohn, N.J.; Tenenholz, B.; Ladda, R.L.; Fink, J.M. Duplication of the distal long arm of chromosome 15: Report of three new patients and review of the literature. Am. J. Med Genet. 2004, 126A, 398–402. [Google Scholar] [CrossRef]
- Hu, X.; Li, L.; Zhang, H.; Hu, Z.; Li, L.; Sun, M.; Liu, R. Prenatal diagnosis of a de novo tetrasomy 15q24.3-25.3: Case report and literature review. J. Clin. Lab. Anal. 2020, 34, e23288. [Google Scholar] [CrossRef]
- Xu, H.; Xiao, B.; Ji, X.; Hu, Q.; Chen, Y.; Qiu, W. Nonmosaic tetrasomy 15q25.2 → qter identified with SNP microarray in a patient with characteristic facial appearance and review of the literature. Eur. J. Med. Genet. 2014, 57, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Herr, H.M.; Scott, C.I., Jr.; Horton, S.J. De novo interstitial direct duplication of 15q: 46,XY,dir dup(15) (pter leads to q24::q14 leads to q21 X 1::q24 leads to qter). J. Med. Genet. 1983, 20, 473–475. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Meng, Z.; Zhang, L.; Luo, X.; Liu, L.; Chen, M.; Li, X.; Zhao, W.; Liang, L. A rare de novo interstitial duplication of 15q15.3q21.2 in a boy with severe short stature, hypogonadism, global developmental delay and intellectual disability. Mol. Cytogenet. 2016, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Wu, X.; Chen, J.; Wu, Y.; Li, S.; Chen, X.; Zhang, X. Aromatase excess syndrome in a Chinese boy due to a novel duplication at 15q21.2. J. Pediatr. Endocrinol. Metab. 2019, 32, 85–88. [Google Scholar] [CrossRef]
- Sakai, L.Y.; Keene, D.R.; Renard, M.; De Backer, J. FBN1: The disease-causing gene for Marfan syndrome and other genetic disorders. Gene 2016, 591, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Cain, S.A.; McGovern, A.; Baldwin, A.; Baldock, C.; Kielty, C.M. Fibrillin-1 Mutations Causing Weill-Marchesani Syndrome and Acromicric and Geleophysic Dysplasias Disrupt Heparan Sulfate Interactions. PLoS ONE 2012, 7, e48634. [Google Scholar] [CrossRef]
- Sakai, L.Y.; Keene, D.R. Fibrillin protein pleiotropy: Acromelic dysplasias. Matrix Biol. 2019, 80, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.S.; Song, H.Y.; Cho, S.Y.; Ki, C.-S.; Yang, S.H.; Kim, O.-H.; Kim, S.J. Acromicric Dysplasia Caused by a Novel Heterozygous Mutation of FBN1 and Effects of Growth Hormone Treatment. Ann. Lab. Med. 2017, 37, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Hulur, I.; Hermanns, P.; Nestoris, C.; Heger, S.; Refetoff, S.; Pohlenz, J.; Grasberger, H. A Single Copy of the Recently Identified Dual Oxidase Maturation Factor (DUOXA) 1 Gene Produces Only Mild Transient Hypothyroidism in a Patient with a Novel Biallelic DUOXA2 Mutation and Monoallelic DUOXA1 Deletion. J. Clin. Endocrinol. Metab. 2011, 96, E841–E845. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Nakajima, M.; Yokoi, T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005, 227, 115–124. [Google Scholar] [CrossRef]
- Losa, M.; Mortini, P.; Pagnano, A.; Detomas, M.; Cassarino, M.F.; Giraldi, F.P. Clinical characteristics and surgical outcome in USP8-mutated human adrenocorticotropic hormone-secreting pituitary adenomas. Endocrine 2019, 63, 240–246. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ke, X.; Yang, H.; Pan, H.; Jiang, Y.; Li, M.; Zhang, H.; Hao, N.; Zhu, H. The Application of Optical Genome Mapping (OGM) in Severe Short Stature Caused by Duplication of 15q14q21.3. Genes 2023, 14, 1016. https://doi.org/10.3390/genes14051016
Ke X, Yang H, Pan H, Jiang Y, Li M, Zhang H, Hao N, Zhu H. The Application of Optical Genome Mapping (OGM) in Severe Short Stature Caused by Duplication of 15q14q21.3. Genes. 2023; 14(5):1016. https://doi.org/10.3390/genes14051016
Chicago/Turabian StyleKe, Xiaoan, Hongbo Yang, Hui Pan, Yulin Jiang, Mengmeng Li, Hanzhe Zhang, Na Hao, and Huijuan Zhu. 2023. "The Application of Optical Genome Mapping (OGM) in Severe Short Stature Caused by Duplication of 15q14q21.3" Genes 14, no. 5: 1016. https://doi.org/10.3390/genes14051016