
Metformin Resistance Is Associated with Expression of Inflammatory and Invasive Genes in A549 Lung Cancer Cells

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Chemicals
2.2. Cell Culture and Viability Assay
2.3. Cell Cycle Analysis
2.4. Western Blot Analysis
2.5. RNA-Seq and Data Analysis
2.6. Reverse Transcription-Quantitative PCR (RT-qPCR)
2.7. Cell Migration Assay
2.8. Immunofluoresce Assay
2.9. Statistics
3. Results
3.1. Establishment of A549-R Cells
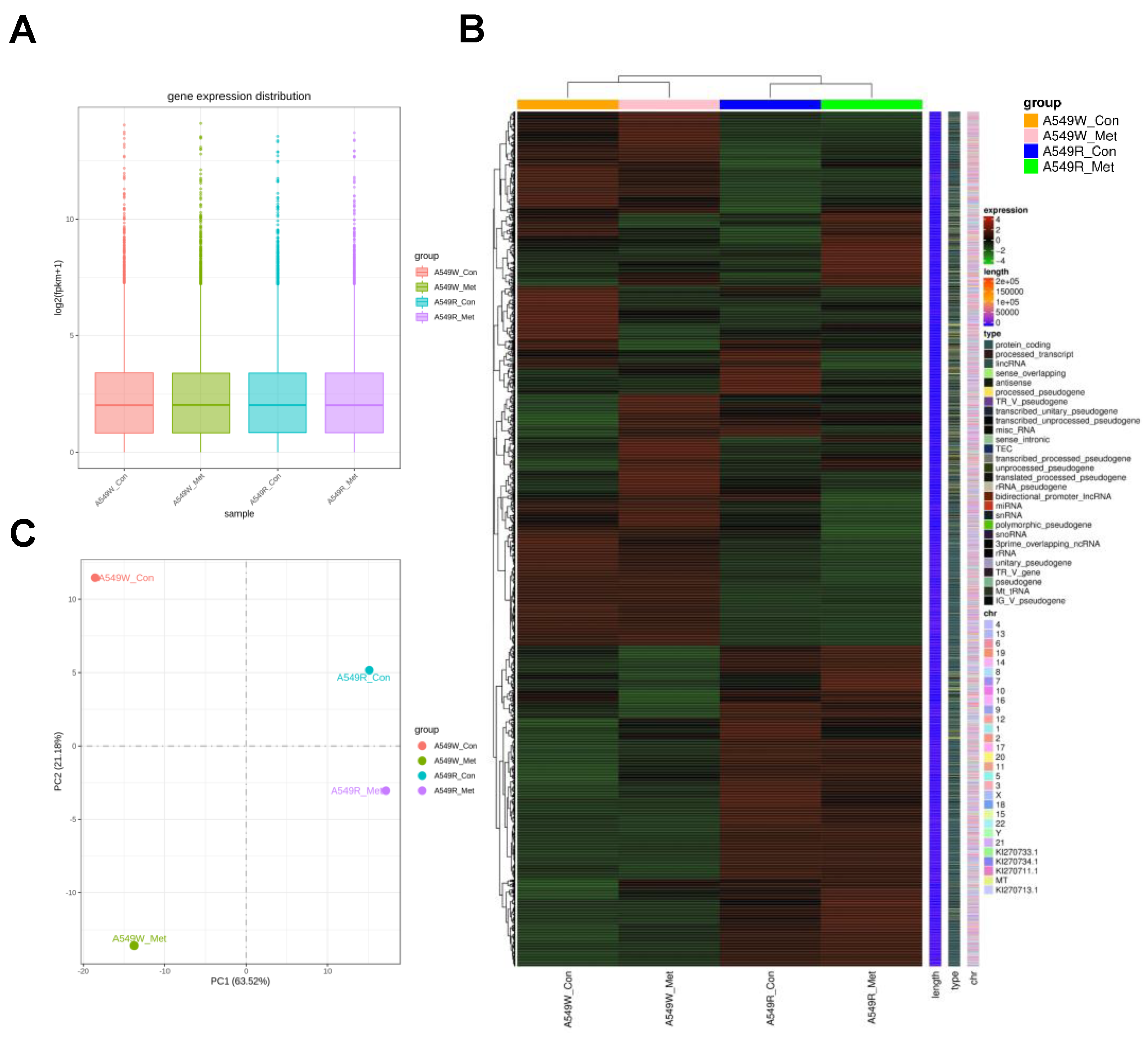
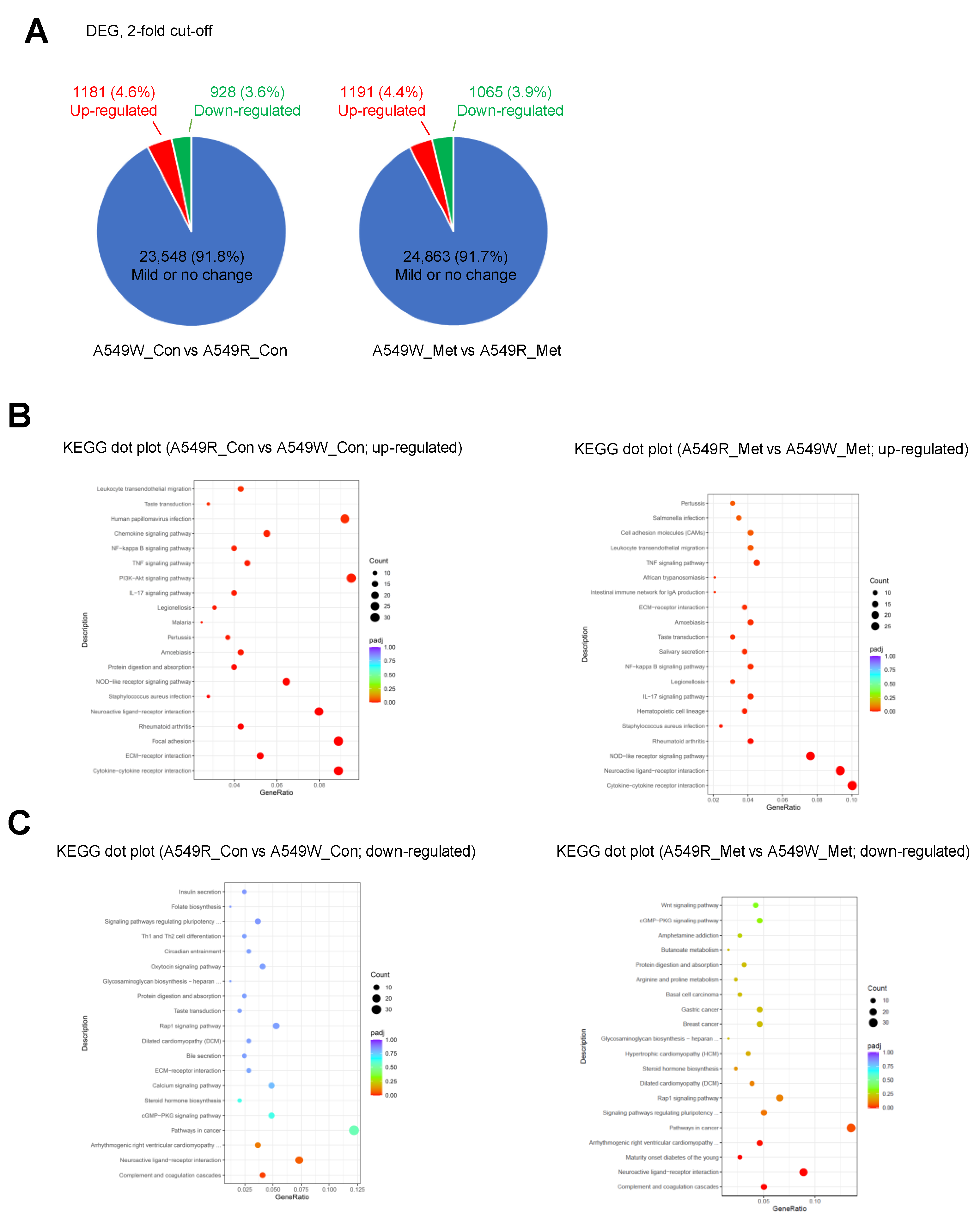
3.2. Gene Expression Profile of A549-R upon Metformin Treatment
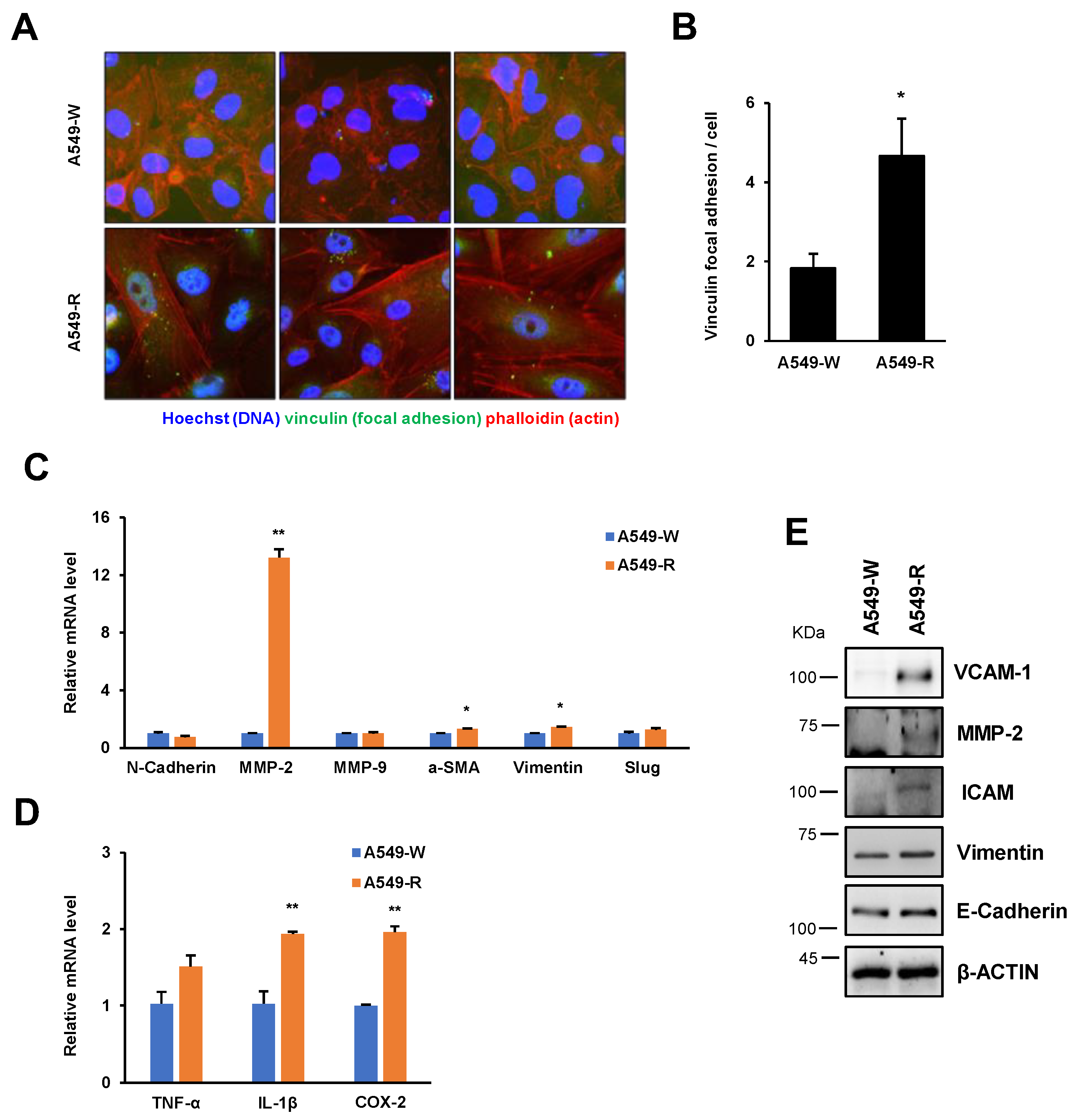
3.3. The Expression of Proinflammatory and Cell Adhesion Genes Is Highly Induced in A549-R
3.4. A549-R Cells Show Various Cancer Cell Phenotype Conversions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Bailey, C.J.; Day, C. Traditional plant medicines as treatments for diabetes. Diabetes Care 1989, 12, 553–564. [Google Scholar] [CrossRef]
- Cicero, A.F.; Tartagni, E.; Ertek, S. Metformin and its clinical use: New insights for an old drug in clinical practice. Arch. Med. Sci. 2012, 8, 907–917. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Tian, X.; Zhang, B.; Li, M.; Wang, Y.; Yang, C.; Wu, J.; Wei, X.; Qu, Q.; Yu, Y.; et al. Low-dose metformin targets the lysosomal AMPK pathway through PEN2. Nature 2022, 603, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013, 494, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Day, E.A.; Ford, R.J.; Smith, B.K.; Mohammadi-Shemirani, P.; Morrow, M.R.; Gutgesell, R.M.; Lu, R.; Raphenya, A.R.; Kabiri, M.; McArthur, A.G.; et al. Metformin-induced increases in GDF15 are important for suppressing appetite and promoting weight loss. Nat. Metab. 2019, 1, 1202–1208. [Google Scholar] [CrossRef]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. 2020, 32, 44–55 e46. [Google Scholar] [CrossRef]
- Pernicova, I.; Korbonits, M. Metformin--mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143–156. [Google Scholar] [CrossRef]
- Giovannucci, E.; Harlan, D.M.; Archer, M.C.; Bergenstal, R.M.; Gapstur, S.M.; Habel, L.A.; Pollak, M.; Regensteiner, J.G.; Yee, D. Diabetes and cancer: A consensus report. Diabetes Care 2010, 33, 1674–1685. [Google Scholar] [CrossRef]
- Leng, W.; Jiang, J.; Chen, B.; Wu, Q. Metformin and Malignant Tumors: Not Over the Hill. Diabetes Metab. Syndr. Obes. 2021, 14, 3673–3689. [Google Scholar] [CrossRef]
- Chan, A.T. Metformin for cancer prevention: A reason for optimism. Lancet Oncol. 2016, 17, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Tossetta, G. Metformin Improves Ovarian Cancer Sensitivity to Paclitaxel and Platinum-Based Drugs: A Review of In Vitro Findings. Int. J. Mol. Sci. 2022, 23, 12893. [Google Scholar] [CrossRef]
- Sui, X.; Xu, Y.; Wang, X.; Han, W.; Pan, H.; Xiao, M. Metformin: A Novel but Controversial Drug in Cancer Prevention and Treatment. Mol. Pharm. 2015, 12, 3783–3791. [Google Scholar] [CrossRef]
- Skuli, S.J.; Alomari, S.; Gaitsch, H.; Bakayoko, A.; Skuli, N.; Tyler, B.M. Metformin and Cancer, an Ambiguanidous Relationship. Pharmaceuticals 2022, 15, 626. [Google Scholar] [CrossRef] [PubMed]
- Joo, S.; Baek, S.; Kang, J.; Seo, D.S.; Kwon, T.K.; Jang, Y. alpha-ketoglutarate suppresses immediate early gene expression in cancer cells. Biochem. Biophys. Res. Commun. 2022, 637, 144–152. [Google Scholar] [CrossRef]
- Wei, M.; Mao, S.; Lu, G.; Li, L.; Lan, X.; Huang, Z.; Chen, Y.; Zhao, M.; Zhao, Y.; Xia, Q. Valproic acid sensitizes metformin-resistant human renal cell carcinoma cells by upregulating H3 acetylation and EMT reversal. BMC Cancer 2018, 18, 434. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jiang, L.; Wang, Y.; Zhao, Y.; Zhang, X.J.; Wu, G.; Zhou, X.; Sun, J.; Bai, J.; Ren, B.; et al. Combination of Metformin and Gefitinib as First-Line Therapy for Nondiabetic Advanced NSCLC Patients with EGFR Mutations: A Randomized, Double-Blind Phase II Trial. Clin. Cancer Res. 2019, 25, 6967–6975. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005, 310, 1642–1646. [Google Scholar] [CrossRef]
- Geiger, B.; Spatz, J.P.; Bershadsky, A.D. Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell. Biol. 2009, 10, 21–33. [Google Scholar] [CrossRef]
- Slattum, G.M.; Rosenblatt, J. Tumour cell invasion: An emerging role for basal epithelial cell extrusion. Nat. Rev. Cancer 2014, 14, 495–501. [Google Scholar] [CrossRef]
- Jiang, S.; Teague, A.M.; Tryggestad, J.B.; Jensen, M.E.; Chernausek, S.D. Role of metformin in epigenetic regulation of placental mitochondrial biogenesis in maternal diabetes. Sci. Rep. 2020, 10, 8314. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Yi, Y.; Liu, Y.; Liu, X.; Keller, E.T.; Qian, C.N.; Zhang, J.; Lu, Y. Metformin targets multiple signaling pathways in cancer. Chin. J. Cancer 2017, 36, 17. [Google Scholar] [CrossRef] [PubMed]
- Chhouri, H.; Alexandre, D.; Grumolato, L. Mechanisms of Acquired Resistance and Tolerance to EGFR Targeted Therapy in Non-Small Cell Lung Cancer. Cancers 2023, 15, 504. [Google Scholar] [CrossRef]
- Al Hilli, M.M.; Bakkum-Gamez, J.N.; Mariani, A.; Cliby, W.A.; Mc Gree, M.E.; Weaver, A.L.; Dowdy, S.C.; Podratz, K.C. The effect of diabetes and metformin on clinical outcomes is negligible in risk-adjusted endometrial cancer cohorts. Gynecol. Oncol. 2016, 140, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Goossens, M.E.; Buntinx, F.; Zeegers, M.P.; Driessen, J.H.; De Bruin, M.L.; De Vries, F. Influence of metformin intake on the risk of bladder cancer in type 2 diabetes patients. Br. J. Clin. Pharmacol. 2015, 80, 1464–1472. [Google Scholar] [CrossRef]
- Becker, C.; Jick, S.S.; Meier, C.R.; Bodmer, M. No evidence for a decreased risk of thyroid cancer in association with use of metformin or other antidiabetic drugs: A case-control study. BMC Cancer 2015, 15, 719. [Google Scholar] [CrossRef]
- Sakoda, L.C.; Ferrara, A.; Achacoso, N.S.; Peng, T.; Ehrlich, S.F.; Quesenberry, C.P., Jr.; Habel, L.A. Metformin use and lung cancer risk in patients with diabetes. Cancer Prev. Res. 2015, 8, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.F.; Zhang, X.L.; Luo, Z.G.; Yan, J.J.; Pan, S.H.; Ying, X.R.; Pan, J.G.; Zhang, G.F. Metformin therapy and prostate cancer risk: A meta-analysis of observational studies. Int. J. Clin. Exp. Med. 2015, 8, 13089–13098. [Google Scholar] [PubMed]
- Oliveras-Ferraros, C.; Vazquez-Martin, A.; Cuyas, E.; Corominas-Faja, B.; Rodriguez-Gallego, E.; Fernandez-Arroyo, S.; Martin-Castillo, B.; Joven, J.; Menendez, J.A. Acquired resistance to metformin in breast cancer cells triggers transcriptome reprogramming toward a degradome-related metastatic stem-like profile. Cell Cycle 2014, 13, 1132–1144. [Google Scholar] [CrossRef]
- Ikediobi, O.N.; Davies, H.; Bignell, G.; Edkins, S.; Stevens, C.; O’Meara, S.; Santarius, T.; Avis, T.; Barthorpe, S.; Brackenbury, L.; et al. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol. Cancer Ther. 2006, 5, 2606–2612. [Google Scholar] [CrossRef]
- Feng, J.; Wang, X.; Ye, X.; Ares, I.; Lopez-Torres, B.; Martinez, M.; Martinez-Larranaga, M.R.; Wang, X.; Anadon, A.; Martinez, M.A. Mitochondria as an important target of metformin: The mechanism of action, toxic and side effects, and new therapeutic applications. Pharmacol. Res. 2022, 177, 106114. [Google Scholar] [CrossRef] [PubMed]
- Fatehi, R.; Rashedinia, M.; Akbarizadeh, A.R.; Zamani, M.; Firouzabadi, N. Metformin enhances anti-cancer properties of resveratrol in MCF-7 breast cancer cells via induction of apoptosis, autophagy and alteration in cell cycle distribution. Biochem. Biophys. Res. Commun. 2023, 644, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Xiao, J.; Liu, J.; Liu, J.; Shu, G.; Yin, G. Metformin suppresses the growth of colorectal cancer by targeting INHBA to inhibit TGF-beta/PI3K/AKT signaling transduction. Cell Death Dis. 2022, 13, 202. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-beta-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [PubMed]
- Lim, Z.F.; Ma, P.C. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol. 2019, 12, 134. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 569–589. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef]
- Hwang, S.H.; Kim, M.C.; Ji, S.; Yang, Y.; Jeong, Y.; Kim, Y. Glucose starvation induces resistance to metformin through the elevation of mitochondrial multidrug resistance protein 1. Cancer Sci. 2019, 110, 1256–1267. [Google Scholar] [CrossRef] [PubMed]
- Zhong, T.; Men, Y.; Lu, L.; Geng, T.; Zhou, J.; Mitsuhashi, A.; Shozu, M.; Maihle, N.J.; Carmichael, G.G.; Taylor, H.S.; et al. Metformin alters DNA methylation genome-wide via the H19/SAHH axis. Oncogene 2017, 36, 2345–2354. [Google Scholar] [CrossRef] [PubMed]
- Cuyas, E.; Fernandez-Arroyo, S.; Verdura, S.; Garcia, R.A.; Stursa, J.; Werner, L.; Blanco-Gonzalez, E.; Montes-Bayon, M.; Joven, J.; Viollet, B.; et al. Metformin regulates global DNA methylation via mitochondrial one-carbon metabolism. Oncogene 2018, 37, 963–970. [Google Scholar] [CrossRef]
- Li, K.; Zhang, T.T.; Wang, F.; Cui, B.; Zhao, C.X.; Yu, J.J.; Lv, X.X.; Zhang, X.W.; Yang, Z.N.; Huang, B.; et al. Metformin suppresses melanoma progression by inhibiting KAT5-mediated SMAD3 acetylation, transcriptional activity and TRIB3 expression. Oncogene 2018, 37, 2967–2981. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Cho, Y.; Chang, M.; Park, S.; Kwon, H. Metformin Decreases 2-HG Production through the MYC-PHGDH Pathway in Suppressing Breast Cancer Cell Proliferation. Metabolites 2021, 11, 480. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, Y.; Lee, B.B.; Cho, E.Y.; Han, J.; Shim, Y.M.; Kim, D.H. Metformin Reduces Histone H3K4me3 at the Promoter Regions of Positive Cell Cycle Regulatory Genes in Lung Cancer Cells. Cancers 2021, 13, 739. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, D.S.; Joo, S.; Baek, S.; Kang, J.; Kwon, T.K.; Jang, Y. Metformin Resistance Is Associated with Expression of Inflammatory and Invasive Genes in A549 Lung Cancer Cells. Genes 2023, 14, 1014. https://doi.org/10.3390/genes14051014
Seo DS, Joo S, Baek S, Kang J, Kwon TK, Jang Y. Metformin Resistance Is Associated with Expression of Inflammatory and Invasive Genes in A549 Lung Cancer Cells. Genes. 2023; 14(5):1014. https://doi.org/10.3390/genes14051014
Chicago/Turabian StyleSeo, Dong Soo, Sungmin Joo, Seungwoo Baek, Jaehyeon Kang, Taeg Kyu Kwon, and Younghoon Jang. 2023. "Metformin Resistance Is Associated with Expression of Inflammatory and Invasive Genes in A549 Lung Cancer Cells" Genes 14, no. 5: 1014. https://doi.org/10.3390/genes14051014