
Mitochondrial Heteroplasmy and PCR Amplification Bias Lead to Wrong Species Delimitation with High Confidence in the South American and Antarctic Marine Bivalve Aequiyoldia eightsii Species Complex

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Collection
2.2. Genomic DNA Extraction and Amplification
2.3. RNA Extraction and cDNA Library Preparation
2.4. De Novo Assembly and SNP Analysis of RNA-Seq Data
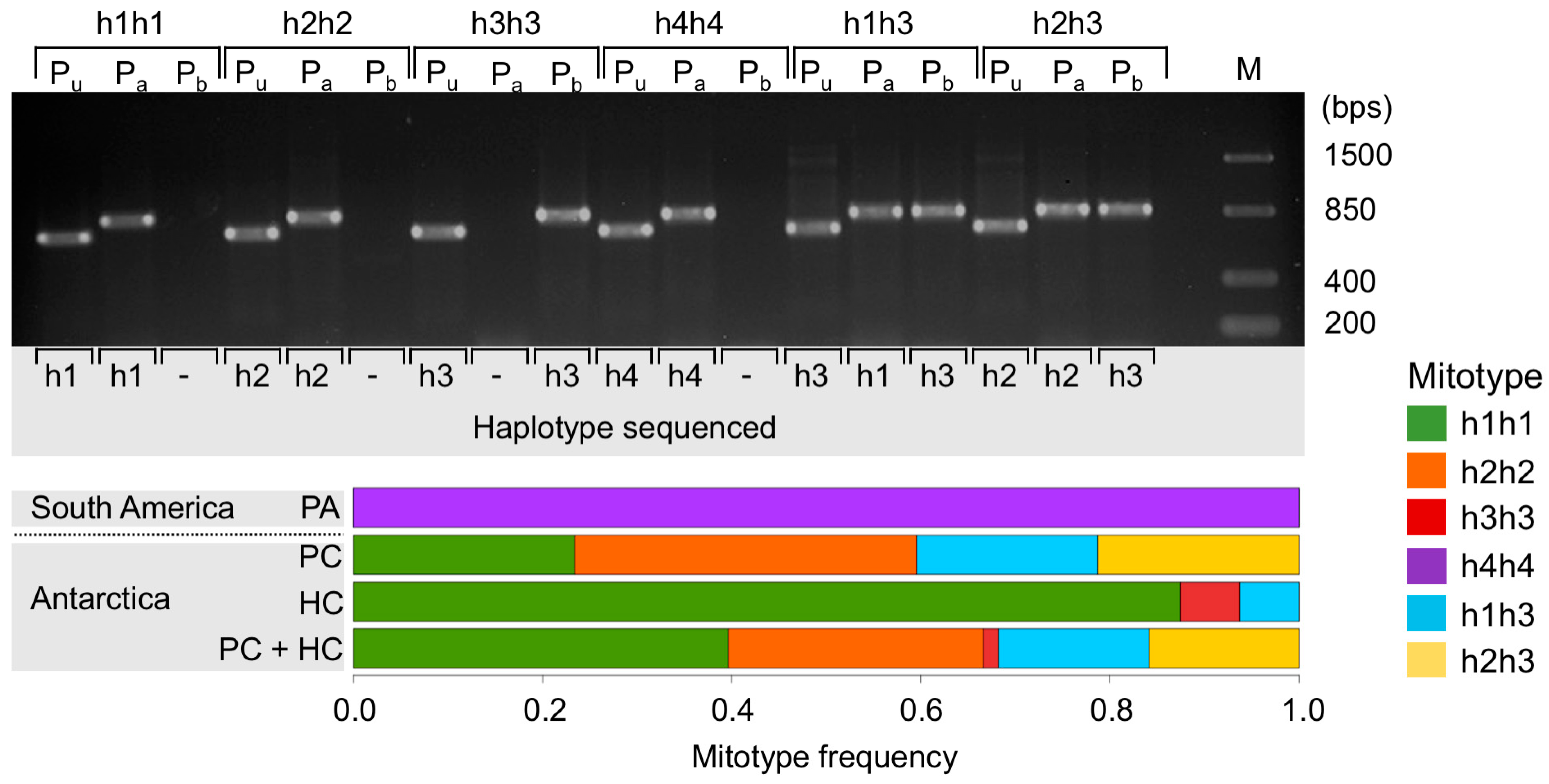
2.5. Coexistence of Mitochondrial Variants in a Single Individual
3. Results
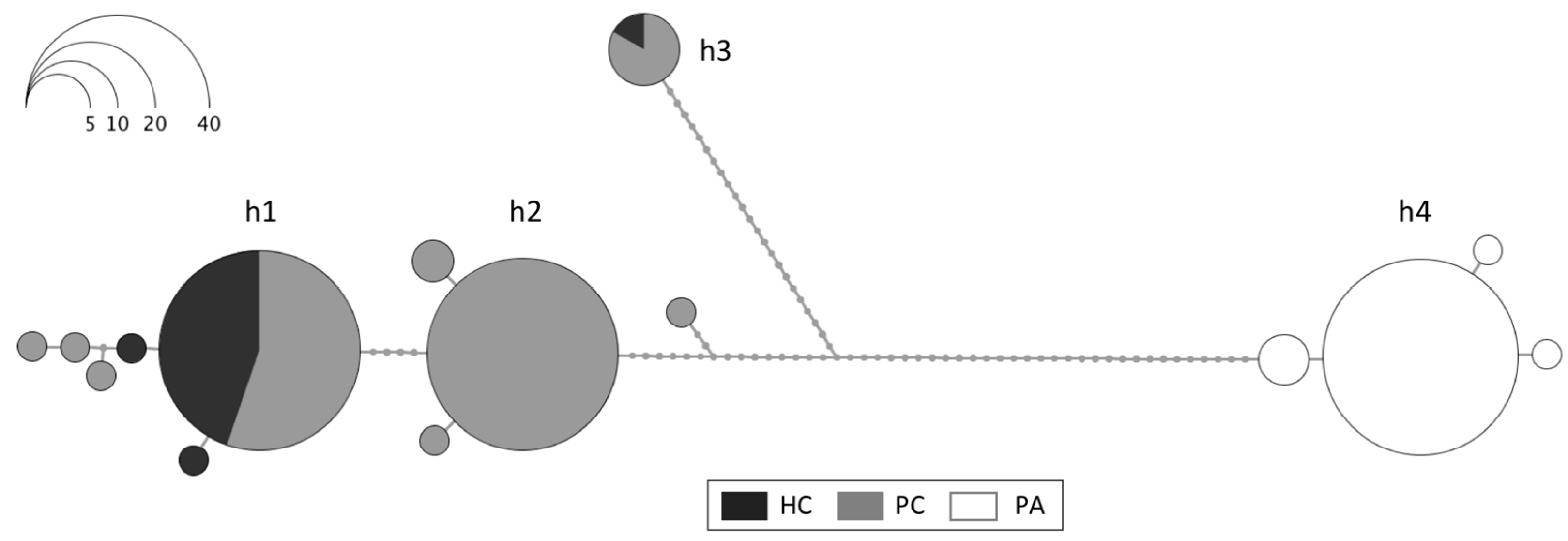
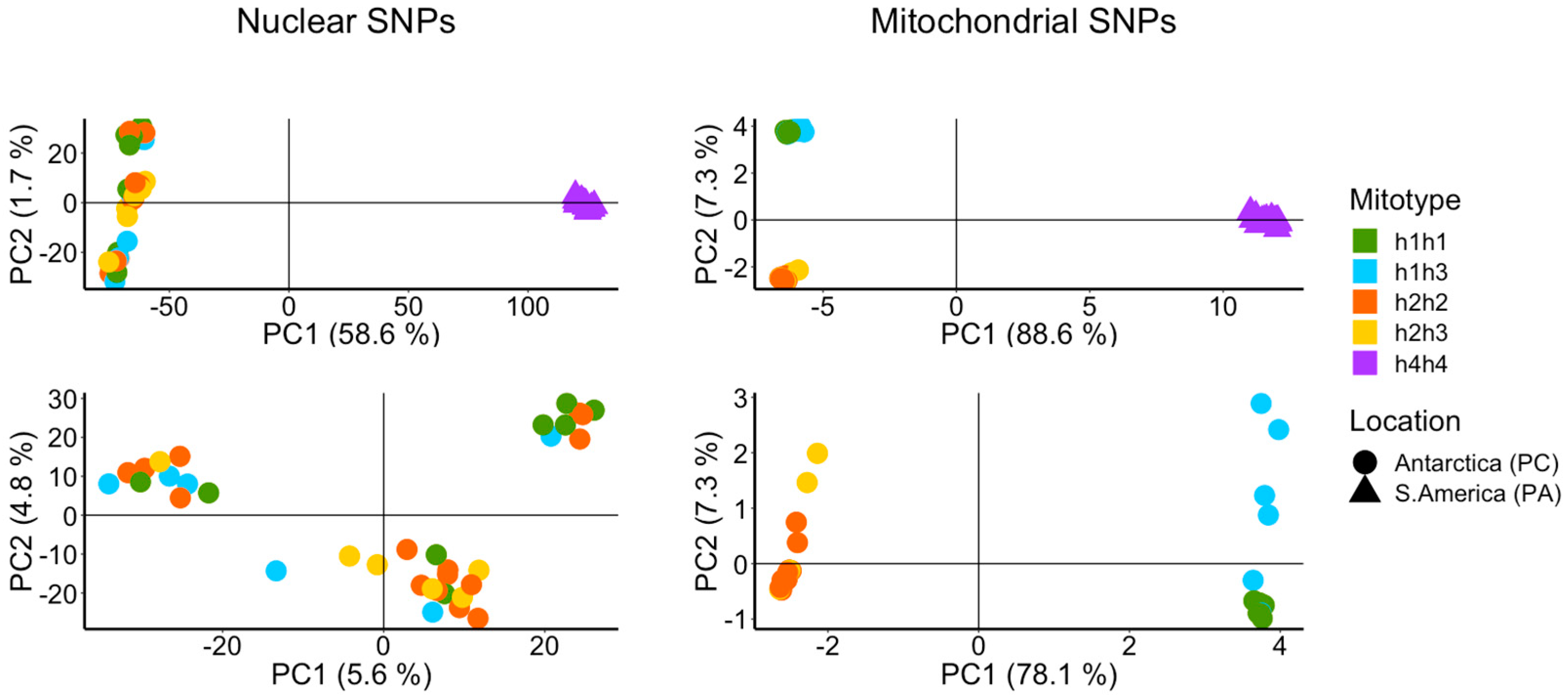
3.1. Mitochondrial and Nuclear Sequence Analysis
3.2. Coexistence of Mitochondrial Variants in a Single Organism
3.3. De Novo Assembly and SNP Analysis
4. Discussion
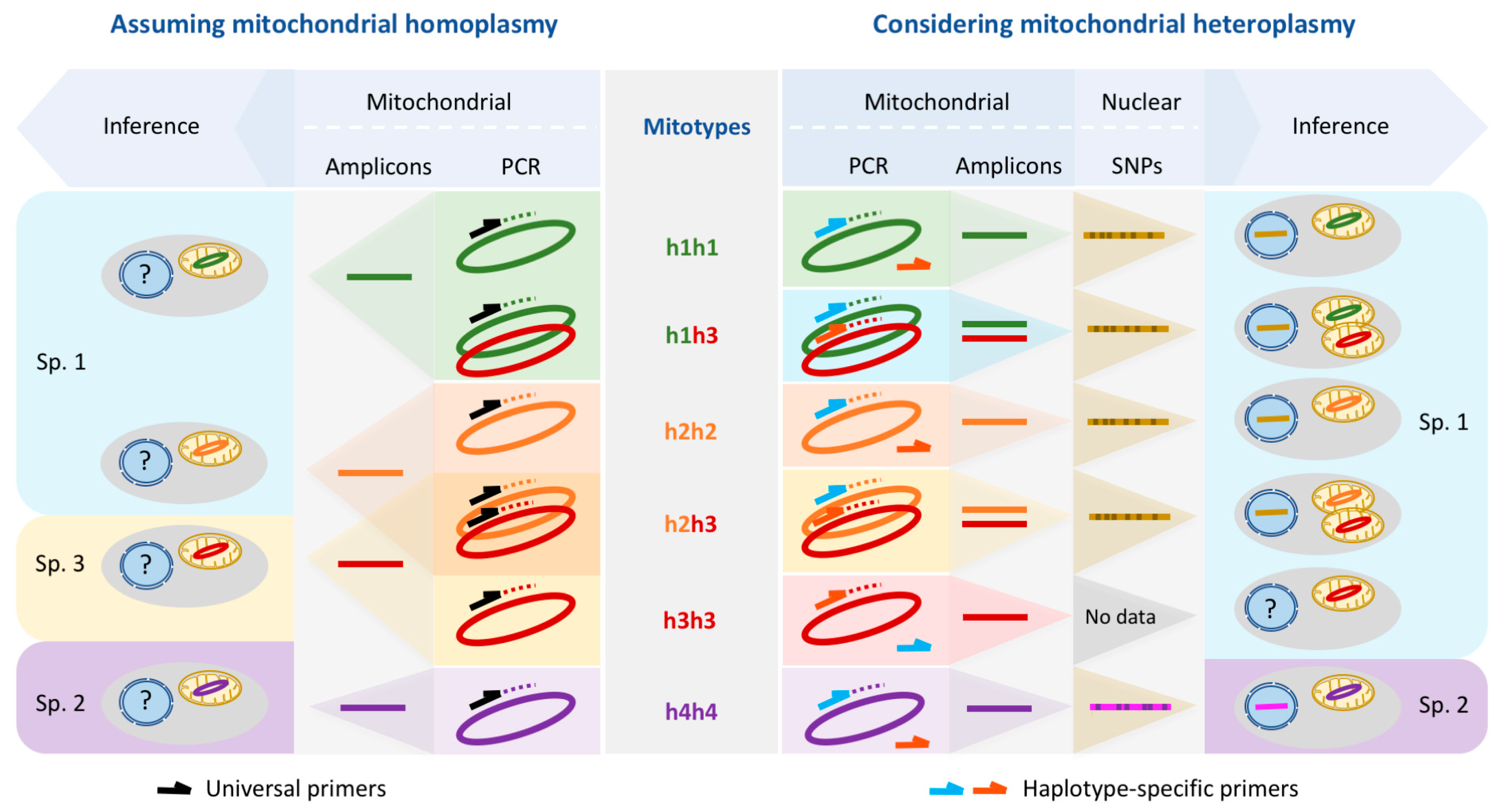
4.1. Incongruence of Methods
4.1.1. Comparison across the Drake Passage
4.1.2. Comparison within the Southern Ocean
4.1.3. Taxonomic Consequences
4.2. True Process: High Mitochondrial Genetic Diversity and Heteroplasmy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; deWaard, J.R. Biological identifications through DNA Barcodes. Proc. Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed]
- DeSalle, R.; Goldstein, P. Review and interpretation of trends in DNA Barcoding. Front. Ecol. Evol. 2019, 7, 302. [Google Scholar] [CrossRef]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA Primers for amplification of mitochondrial Cytochrome c Oxidase Subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [PubMed]
- Steinke, D.; Prosser, S.W.J.; Hebert, P.D.N. DNA Barcoding of marine metazoans. In Marine Genomics; Springer: Berlin/Heidelberg, Germany, 2016; pp. 155–168. [Google Scholar]
- Rollins, L.A.; Woolnough, A.P.; Fanson, B.G.; Cummins, M.L.; Crowley, T.M.; Wilton, A.N.; Sinclair, R.; Butler, A.; Sherwin, W.B. Selection on mitochondrial variants occurs between and within individuals in an expanding invasion. Mol. Biol. Evol. 2016, 33, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Ratnasingham, S.; Hebert, P.D. BOLD: The Barcode of Life Data System (Http://Www. Barcodinglife. Org). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef]
- Françoso, E.; Zuntini, A.R.; Ricardo, P.C.; Silva, J.P.N.; Brito, R.; Oldroyd, B.P.; Arias, M.C. Conserved Numts mask a highly divergent mitochondrial-COI gene in a species complex of Australian stingless bees Tetragonula (Hymenoptera: Apidae). Mitochondrial DNA Part A 2019, 30, 806–817. [Google Scholar] [CrossRef]
- Kim, S.-J.; Lee, K.Y.; Ju, S.-J. Nuclear mitochondrial pseudogenes in Austinograea alayseae hydrothermal vent crabs (Crustacea: Bythograeidae): Effects on DNA Barcoding. Mol. Ecol. Resour. 2013, 13, 781–787. [Google Scholar] [CrossRef]
- Iketani, G.; Pimentel, L.; Torres, E.D.S.; do Rêgo, P.S.; Sampaio, I. Mitochondrial heteroplasmy and pseudogenes in the Freshwater prawn, Macrobrachium amazonicum (Heller, 1862): DNA Barcoding and phylogeographic implications. Mitochondrial. DNA Part A 2021, 32, 1–11. [Google Scholar] [CrossRef]
- Mastrantonio, V.; Latrofa, M.S.; Porretta, D.; Lia, R.P.; Parisi, A.; Iatta, R.; Dantas-Torres, F.; Otranto, D.; Urbanelli, S. Paternal leakage and mtDNA heteroplasmy in Rhipicephalus spp. Ticks. Sci. Rep. 2019, 9, 1460. [Google Scholar] [CrossRef]
- Dégletagne, C.; Abele, D.; Held, C. A Distinct mitochondrial genome with DUI-Like inheritance in the ocean quahog Arctica islandica. Mol. Biol. Evol. 2016, 33, 375–383. [Google Scholar] [CrossRef]
- Doublet, V.; Souty-Grosset, C.; Bouchon, D.; Cordaux, R.; Marcadé, I. A thirty million year-old inherited heteroplasmy. PLoS ONE 2008, 3, e2938. [Google Scholar] [CrossRef]
- Bentzen, P.; Leggett, W.C.; Brown, G.G. Length and restriction site heteroplasmy in the mitochondrial DNA of american shad (Alosa sapidissima). Genetics 1988, 118, 509–518. [Google Scholar] [CrossRef]
- Kvist, L.; Martens, J.; Nazarenko, A.A.; Orell, M. Paternal leakage of mitochondrial DNA in the great tit (Parus major). Mol. Biol. Evol. 2003, 20, 243–247. [Google Scholar] [CrossRef]
- Vollmer, N.L.; Viricel, A.; Wilcox, L.; Katherine Moore, M.; Rosel, P.E. The Occurrence of mtDNA heteroplasmy in multiple cetacean species. Curr. Genet. 2011, 57, 115–131. [Google Scholar] [CrossRef]
- Ghiselli, F.; Maurizii, M.G.; Reunov, A.; Ariño-Bassols, H.; Cifaldi, C.; Pecci, A.; Alexandrova, Y.; Bettini, S.; Passamonti, M.; Franceschini, V.; et al. Natural heteroplasmy and mitochondrial inheritance in bivalve molluscs. Integr. Comp. Biol. 2019, 59, 1016–1032. [Google Scholar] [CrossRef]
- Gusman, A.; Lecomte, S.; Stewart, D.T.; Passamonti, M.; Breton, S. Pursuing the quest for better understanding the taxonomic distribution of the system of Doubly Uniparental Inheritance of mtDNA. PeerJ 2016, 4, e2760. [Google Scholar] [CrossRef]
- Zouros, E.; Oberhauser Ball, A.; Saavedra, C.; Freeman, K.R. An unusual type of mitochondrial DNA inheritance in the blue mussel Mytilus. Proc. Natl. Acad. Sci. USA 1994, 91, 7463–7467. [Google Scholar] [CrossRef]
- Zouros, E.; Rodakis, G.C. Doubly Uniparental Inheritance of mtDNA: An unappreciated defiance of a general rule. In Cellular and Molecular Basis of Mitochondrial Inheritance; Springer: Berlin/Heidelberg, Germany, 2019; Volume 231, pp. 25–49. [Google Scholar] [CrossRef]
- Doucet-Beaupré, H.; Breton, S.; Chapman, E.G.; Blier, P.U.; Bogan, A.E.; Stewart, D.T.; Hoeh, W.R. Mitochondrial phylogenomics of the bivalvia (Mollusca): Searching for the origin and mitogenomic correlates of Doubly Uniparental Inheritance of mtDNA. BMC Evol. Biol. 2010, 10, 50. [Google Scholar] [CrossRef]
- Terranova, M.S.; Lo Brutto, S.; Arculeo, M.; Mitton, J.B. A Mitochondrial phylogeography of Brachidontes variabilis (Bivalvia: Mytilidae) reveals three cryptic species. J. Zool. Syst. Evol. Res. 2007, 45, 289–298. [Google Scholar] [CrossRef]
- Boyle, E.E.; Etter, R.J. Heteroplasmy in a deep-Sea protobranch bivalve suggests an ancient origin of Doubly Uniparental Inheritance of mitochondria in bivalvia. Mar. Biol. 2012, 160, 413–422. [Google Scholar] [CrossRef]
- Sharma, P.P.; Zardus, J.D.; Boyle, E.E.; González, V.L.; Jennings, R.M.; McIntyre, E.; Wheeler, W.C.; Etter, R.J.; Giribet, G. Into the Deep: A phylogenetic approach to the bivalve subclass protobranchia. Mol. Phylogenetics Evol. 2013, 69, 188–204. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.J.; Linse, K.; Thatje, S. Differential adaptations between cold-stenothermal environments in the bivalve Lissarca Cf. miliaris (Philobryidae) from the Scotia Sea islands and Antarctic Peninsula. J. Sea Res. 2014, 88, 11–20. [Google Scholar] [CrossRef]
- González-Wevar, C.A.; Díaz, A.; Gérard, K.; Cañete, J.I.; Poulin, E. Divergence time estimations and contrasting patterns of genetic diversity between antarctic and southern South America benthic invertebrates. Rev. Chil. Hist. Nat. 2012, 85, 445–456. [Google Scholar]
- Muñoz-Ramírez, C.P.; Barnes, D.K.A.; Cárdenas, L.; Meredith, M.P.; Morley, S.A.; Roman-Gonzalez, A.; Sands, C.J.; Scourse, J.; Brante, A. Gene flow in the antarctic bivalve Aequiyoldia eightsii (Jay, 1839) suggests a role for the Antarctic Peninsula coastal current in larval dispersal. R. Soc. Open Sci. 2020, 7, 200603. [Google Scholar] [CrossRef]
- Martínez, M.; González-Aravena, M.; Held, C.; Abele, D. A molecular perspective on the invasibility of the Southern Ocean benthos: The impact of hypoxia and temperature on gene expression in South American and Antarctic Aequiyoldia bivalves. Front. Physiol. 2023, 14, 269. [Google Scholar]
- González-Wevar, C.A.; Gérard, K.; Rosenfeld, S.; Saucède, T.; Naretto, J.; Díaz, A.; Morley, S.A.; Brickle, P.; Poulin, E. Cryptic speciation in Southern Ocean Aequiyoldia eightsii (Jay, 1839): Mio-Pliocene trans-Drake Passage separation and diversification. Prog. Oceanogr. 2019, 174, 44–54. [Google Scholar] [CrossRef]
- Low, T.Y.; Mohtar, M.A.; Ang, M.Y.; Jamal, R. Connecting proteomics to Next-Generation Sequencing: Proteogenomics and its current applications in biology. Proteomics 2019, 19, e1800235. [Google Scholar] [CrossRef]
- De Wit, P.; Pespeni, M.H.; Palumbi, S.R. SNP genotyping and population genomics from expressed sequences—Current advances and future possibilities. Mol. Ecol. 2015, 24, 2310–2323. [Google Scholar] [CrossRef]
- García, G.; Ríos, N.; Gutiérrez, V.; Serra, S.; Loureiro, M. Transcriptome-based SNP discovery and validation in the hybrid zone of the neotropical annual fish Genus Austrolebias. Genes 2019, 10, 789. [Google Scholar] [CrossRef]
- Yan, J.; Song, Z.; Xu, Q.; Kang, L.; Zhu, C.; Xing, S.; Liu, W.; Greimler, J.; Züst, T.; Li, J.; et al. Population transcriptomic characterization of the genetic and expression variation of a candidate progenitor of Miscanthus energy Crops. Mol. Ecol. 2017, 26, 5911–5922. [Google Scholar] [CrossRef]
- Rogier, O.; Chateigner, A.; Amanzougarene, S.; Lesage-Descauses, M.-C.; Balzergue, S.; Brunaud, V.; Caius, J.; Soubigou-Taconnat, L.; Jorge, V.; Segura, V. Accuracy of RNAseq based SNP discovery and genotyping in Populus nigra. BMC Genom. 2018, 19, 909. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap. BBMap Short Read Aligner, and Other Bioinformatic Tools. 2014. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 24 July 2021).
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- O’Leary, S.J.; Puritz, J.B.; Willis, S.C.; Hollenbeck, C.M.; Portnoy, D.S. These aren’t the loci you’e looking for: Principles of effective SNP filtering for molecular ecologists. Mol. Ecol. 2018, 27, 3193–3206. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Verity, R.; Nichols, R.A. What is genetic differentiation, and how should we measure It--GST, D, neither or both? Mol. Ecol. 2014, 23, 4216–4225. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of Molecular Variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef]
- RCore, T. R: A Language and Environment for Statistical Computing; R foundation for statistical computing: Vienna, Austria, 2016. [Google Scholar]
- Knaus, B.J.; Grünwald, N.J. Vcfr: A package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 2017, 17, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T.; Ahmed, I. Adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef] [PubMed]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [PubMed]
- Dray, S.; Dufour, A.-B. The Ade4 package: Implementing the duality diagram for ecologists. J. Stat. Softw. 2007, 22, 1–20. [Google Scholar] [CrossRef]
- Paradis, E. Pegas: An R package for population genetics with an integrated–modular approach. Bioinformatics 2010, 26, 419–420. [Google Scholar] [CrossRef]
- Milani, L.; Ghiselli, F. Faraway, so close. The comparative method and the potential of non-model animals in mitochondrial research. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2020, 375, 20190186. [Google Scholar] [CrossRef]
- David, K.T.; Wilson, A.E.; Halanych, K.M. Sequencing disparity in the genomic era. Mol. Biol. Evol. 2019, 36, 1624–1627. [Google Scholar] [CrossRef]
- Dietz, L.; Arango, C.P.; Dömel, J.S.; Halanych, K.M.; Harder, A.M.; Held, C.; Mahon, A.R.; Mayer, C.; Melzer, R.R.; Rouse, G.W.; et al. Regional differentiation and extensive hybridization between mitochondrial clades of the Southern Ocean giant sea spider Colossendeis megalonyx. R. Soc. Open Sci. 2015, 2, 140424. [Google Scholar] [CrossRef]
- Baird, H.P.; Miller, K.J.; Stark, J.S. Evidence of hidden biodiversity, ongoing speciation and diverse patterns of genetic structure in giant antarctic amphipods. Mol. Ecol. 2011, 20, 3439–3454. [Google Scholar] [CrossRef]
- Held, C. Molecular evidence for cryptic speciation within the widespread antarctic crustacean Ceratoserolis trilobitoides (Crustacea, Isopoda). In Antarctic Biology in A Global Context; Gregor Rare Books: Langley, WA, USA, 2003; pp. 135–139. ISBN 90-5782-079-X. [Google Scholar]
- Neal, L.; Wiklund, H.; Muir, A.I.; Linse, K.; Glover, A.G. The identity of juvenile Polynoidae (Annelida) in the Southern Ocean revealed by DNA taxonomy, with notes on the status of Herdmanella gracilis ehlers Sensu augener. Mem. Mus. Vic. 2014, 71, 203–216. [Google Scholar]
- Hemery, L.; Eléaume, M.; Roussel, V.; Améziane, N.; Gallut, C.; Steinke, D.; Cruaud, C.; Couloux, A.; Wilson, N. Comprehensive sampling reveals circumpolarity and sympatry in seven mitochondrial lineages of the Southern Ocean crinoid species Promachocrinus kerguelensis (Echinodermata). Mol. Ecol. 2012, 21, 2502–2518. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source of Variation | df | Sum of Squares | Percentage of Variation | p Value | ||||
|---|---|---|---|---|---|---|---|---|
| Nuc. | Mit. | Nuc. | Mit. | Nuc. | Mit. | Nuc. | Mit. | |
| Between location | 1 | 1 | 4.33 | 10.33 | 67.73 | 89.58 | 0.001 | 0.001 |
| Between mitotype, within location | 3 | 3 | 0.09 | 0.30 | 0.57 | 7.82 | 0.442 | 0.001 |
| Within individual | 49 | 49 | 0.08 | 0.012 | 31.70 | 2.60 | 0.001 | 0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez, M.; Harms, L.; Abele, D.; Held, C. Mitochondrial Heteroplasmy and PCR Amplification Bias Lead to Wrong Species Delimitation with High Confidence in the South American and Antarctic Marine Bivalve Aequiyoldia eightsii Species Complex. Genes 2023, 14, 935. https://doi.org/10.3390/genes14040935
Martínez M, Harms L, Abele D, Held C. Mitochondrial Heteroplasmy and PCR Amplification Bias Lead to Wrong Species Delimitation with High Confidence in the South American and Antarctic Marine Bivalve Aequiyoldia eightsii Species Complex. Genes. 2023; 14(4):935. https://doi.org/10.3390/genes14040935
Chicago/Turabian StyleMartínez, Mariano, Lars Harms, Doris Abele, and Christoph Held. 2023. "Mitochondrial Heteroplasmy and PCR Amplification Bias Lead to Wrong Species Delimitation with High Confidence in the South American and Antarctic Marine Bivalve Aequiyoldia eightsii Species Complex" Genes 14, no. 4: 935. https://doi.org/10.3390/genes14040935