
Identification of GLI1 and KIAA0825 Variants in Two Families with Postaxial Polydactyly
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Approval and Subjects
2.2. Whole-Exome Sequencing and Data Analysis
2.3. Pathogenicity Index and Protein Stability
2.4. Sanger Sequencing
2.5. Modeling and Interaction Studies
3. Results
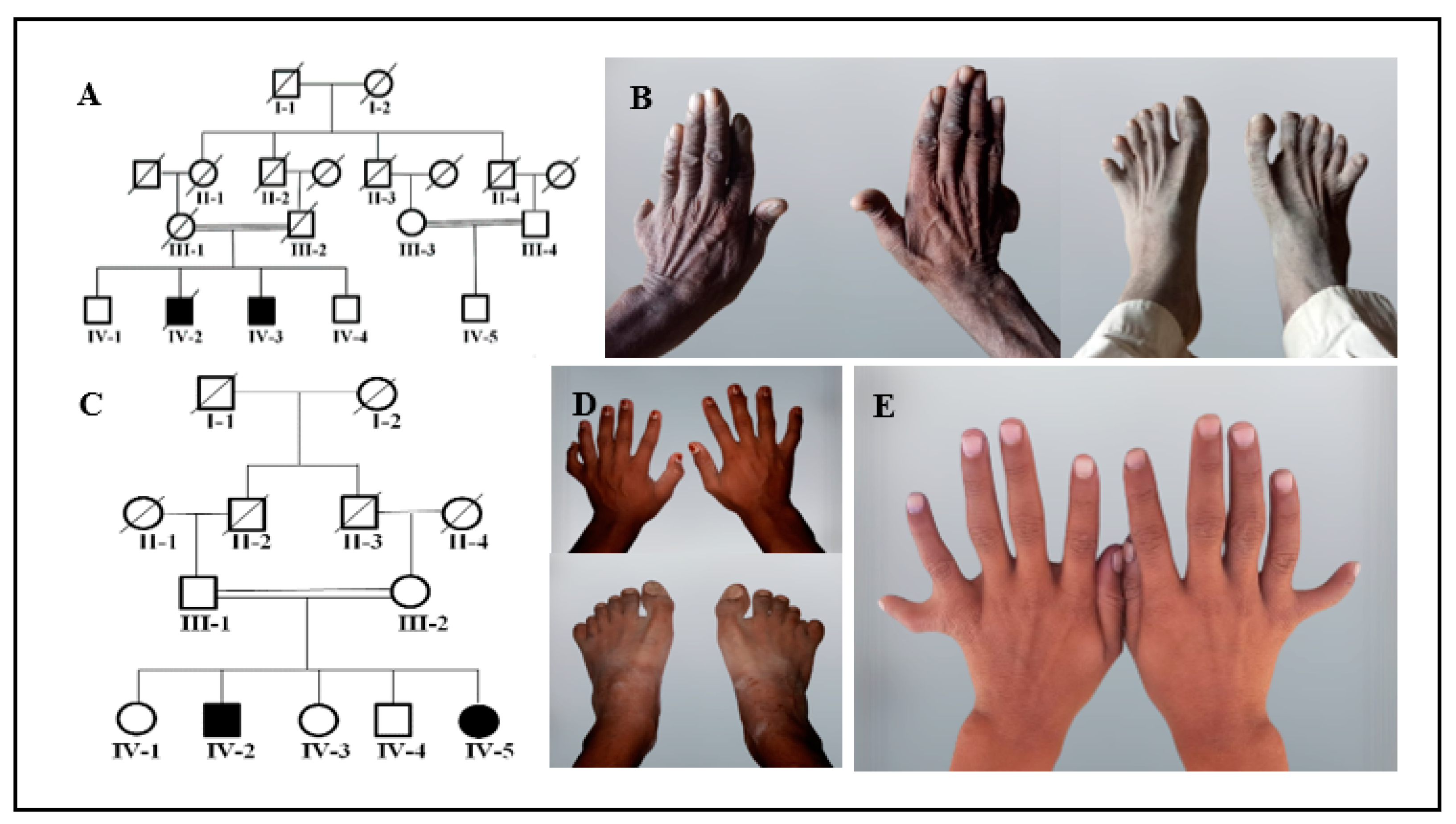
3.1. Clinical Description
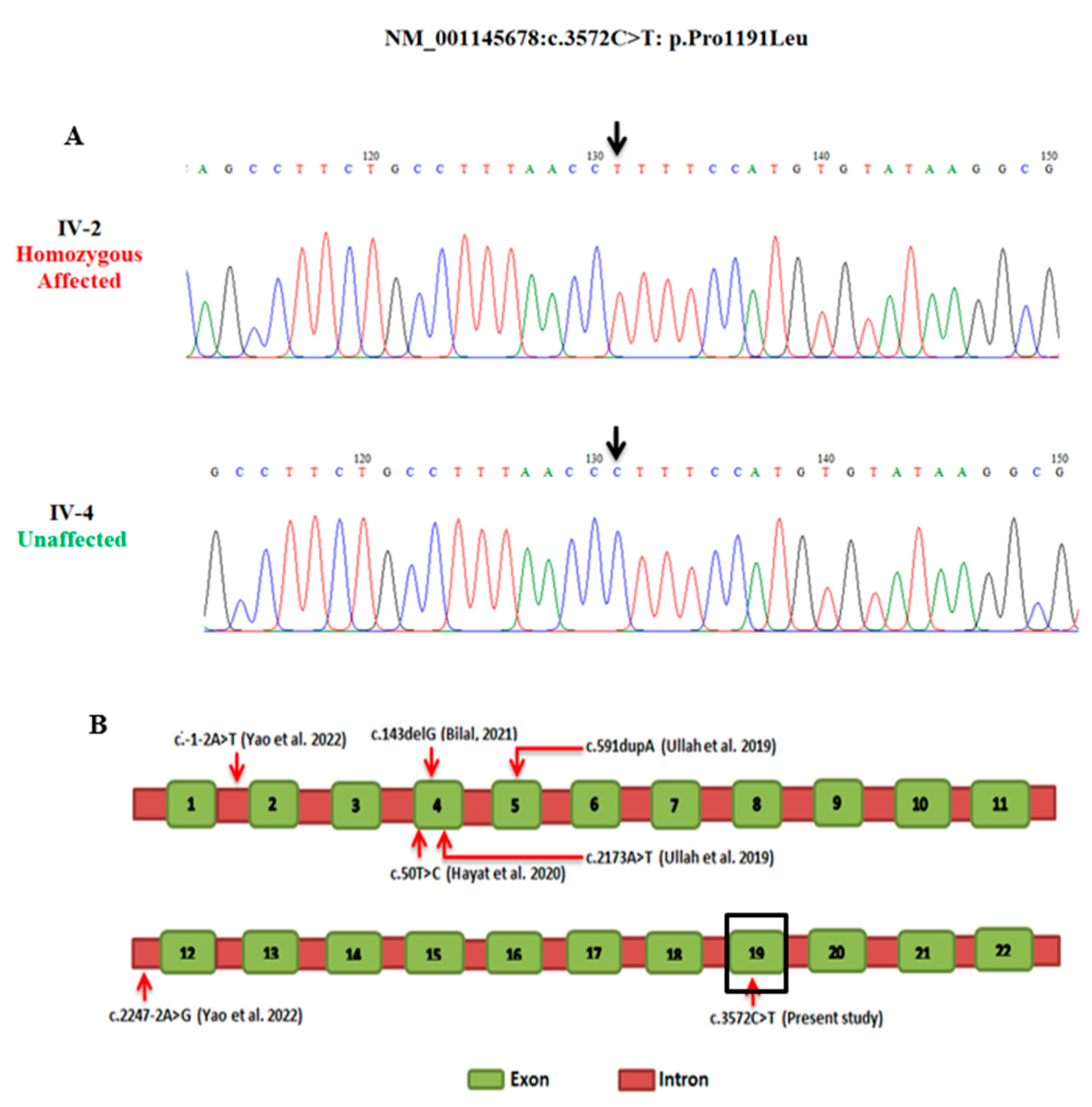
3.2. Molecular Findings
3.3. Pathogenicity Validation and Protein Stability
3.4. Modeling and Docking Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xiang, Y.; Bian, J.; Wang, Z.; Xu, Y.; Fu, Q. Clinical study of 459 polydactyly cases in China, 2010 to 2014. Congenit. Anom. 2016, 56, 226–232. [Google Scholar] [CrossRef]
- Ahmad, S.; Ali, M.Z.; Muzammal, M.; Mir, F.A.; Khan, M.A. The molecular genetics of human appendicular skeleton. Mol. Genet. Genom. 2022, 297, 1195–1214. [Google Scholar] [CrossRef]
- Umair, M.; Ahmad, F.; Bilal, M.; Ahmad, W.; Alfadhel, M. Clinical genetics of polydactyly: An updated review. Front. Genet. 2018, 9, 447. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, Z.; Liaqat, R.; Palander, O.; Bilal, M.; Zeb, S.; Ahmad, F.; Jawad Khan, M.; Umair, M. Genetic overview of postaxial polydactyly: Updated classification. Clin. Genet. 2022, 103, 3–15. [Google Scholar] [CrossRef]
- Biesecker, L.G. Polydactyly: How many disorders and how many genes? 2010 update. Dev. Dyn. 2011, 240, 931–942. [Google Scholar] [CrossRef] [Green Version]
- Malik, S. Polydactyly: Phenotypes, genetics and classification. Clin. Genet. 2014, 85, 203–212. [Google Scholar] [CrossRef]
- Hayat, A.; Umair, M.; Abbas, S.; Rauf, A.; Ahmad, F.; Ullah, S.; Ahmad, W.; Khan, B. Identification of a novel biallelic missense variant in the KIAA0825 underlies postaxial polydactyly type A. Genomics 2020, 112, 2729–2733. [Google Scholar] [CrossRef]
- Bilal, M.; Ahmad, W. A Frameshift Variant in KIAA0825 Causes Postaxial Polydactyly. Mol. Syndromol. 2021, 12, 20–24. [Google Scholar] [CrossRef]
- Stewart, T.A.; Bhat, R.; Newman, S.A. The evolutionary origin of digit patterning. EvoDevo 2017, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Verma, P.K.; El-Harouni, A.A. Review of literature: Genes related to postaxial polydactyly. Front. Pediatr. 2015, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Tickle, C.; Towers, M. Sonic hedgehog signaling in limb development. Front. Cell Dev. Biol. 2017, 5, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Rios, J. The many lives of SHH in limb development and evolution. Semin. Cell Dev. Biol. 2016, 49, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Koscielny, G.; Yaikhom, G.; Iyer, V.; Meehan, T.F.; Morgan, H.; Atienza-Herrero, J.; Blake, A.; Chen, C.-K.; Easty, R.; Di Fenza, A. The International Mouse Phenotyping Consortium Web Portal, a unified point of access for knockout mice and related phenotyping data. Nucleic Acids Res. 2014, 42, D802–D809. [Google Scholar] [CrossRef] [Green Version]
- Steinhaus, R.; Proft, S.; Schuelke, M.; Cooper, D.N.; Schwarz, J.M.; Seelow, D. MutationTaster2021. Nucleic Acids Res. 2021, 49, W446–W451. [Google Scholar] [CrossRef]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice—Improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021, 13, 1–12. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.; Edwards, K.J.; Day, I.N.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2. 0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Randall, A.; Baldi, P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins Struct. Funct. Bioinform. 2006, 62, 1125–1132. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Ugene Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. Available online: http://ugene.net/ (accessed on 5 September 2022).
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Vajda, S.; Yueh, C.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Kozakov, D. New additions to the C lus P ro server motivated by CAPRI. Proteins: Struct. Funct. Bioinform. 2017, 85, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Palencia-Campos, A.; Ullah, A.; Nevado, J.; Yıldırım, R.; Unal, E.; Ciorraga, M.; Barruz, P.; Chico, L.; Piceci-Sparascio, F.; Guida, V. GLI1 inactivation is associated with developmental phenotypes overlapping with Ellis–van Creveld syndrome. Hum. Mol. Genet. 2017, 26, 4556–4571. [Google Scholar] [CrossRef] [Green Version]
- Ullah, I.; Kakar, N.; Schrauwen, I.; Hussain, S.; Chakchouk, I.; Liaqat, K.; Acharya, A.; Wasif, N.; Santos-Cortez, R.L.P.; Khan, S. Variants in KIAA0825 underlie autosomal recessive postaxial polydactyly. Hum. Genet. 2019, 138, 593–600. [Google Scholar] [CrossRef]
- Yao, Y.; Deng, S.; Zhu, F. Prenatal Detection of Novel Compound Heterozygous Splice Site Variants of the KIAA0825 Gene in a Fetus with Postaxial Polydactyly Type A. Genes 2022, 13, 1230. [Google Scholar] [CrossRef] [PubMed]
- Bakar, A.; Ullah, A.; Bibi, N.; Khan, H.; ur Rahman, A.; Ahmad, W.; Khan, B. A novel homozygous variant in the GLI1 underlies postaxial polydactyly in a large consanguineous family with intra familial variable phenotypes. Eur. J. Med. Genet. 2022, 65, 104599. [Google Scholar] [CrossRef] [PubMed]
- Yousaf, M.; Ullah, A.; Azeem, Z.; Isani Majeed, A.; Memon, M.I.; Ghous, T.; Basit, S.; Ahmad, W. Novel heterozygous sequence variant in the GLI1 underlies postaxial polydactyly. Congenit. Anom. 2020, 60, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Umair, M.; Majeed, A.I.; Jan, A.; Ahmad, W. A novel homozygous sequence variant in GLI1 underlies first case of autosomal recessive pre-axial polydactyly. Clin. Genet. 2019, 95, 540–541. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Clinical Examinations | Gene/ Mutation | |||||
|---|---|---|---|---|---|---|---|
| Subject | Polydactyly | Syndactyly | Clinodactyly | Brachydactyly | Other Anomaly | ||
| A | IV-2 (Male) | Bilateral PAPA of hands and bilateral PAPA of feet | Bilateral 2/3 cutaneous syndactyly of feet | Bilateral clinodactyly of hands | − | Sandal gap abnormal big toe shape | KIAA0825 c.3572C>T p.Pro1191Leu |
| IV-3 (Male) | Bilateral PAPA of hands | − | − | − | − | ||
| B | IV-2 (Male) | Unilateral PAPA of hand and bilateral PAPA of feet | − | − | − | − | GLI1 c.337C>T p.Arg113* |
| IV-5 (Female) | Bilateral PAPA of hands | − | − | − | − | ||
| Gene | Zygosity | Genomic Position (hg19) | mRNA Transcript | cDNA Change | Amino Acid Change | gnomAD Allele Count | gnomAD All | gnomAD South Asian | |
|---|---|---|---|---|---|---|---|---|---|
| KIAA0825 | Homozygous | 5:93,721,994 | NM_001145678 | c.3572C>T | p.Pro1191Leu | 9 Heterozygote | 0.00005731 | 0.0003953 | |
| Tools | CADD_phred | Mutation Taster | Mutation Assessor | M-cap | Sift | Polyphen-2 | FATHMM | Provean | |
| Predictions | 33 | Disease causing | Medium | Damaging | Damaging | Damaging | Damaging | Damaging | |
| GLI1 | Zygosity | Genomic position (hg19) | mRNA Transcript | cDNA change | Amino acid change | gnomAD allele count | gnomAD All | gnomAD South Asian | |
| Homozygous | 12:57,858,599 | NM_005269 | c.337C>T | p.Arg113* | 1 Heterozygote | 0.00000397 | 0.0000326 | ||
| Tools | CADD_phred | Mutation Taster | FATHMM | ||||||
| Predictions | 36 | Disease causing | Damaging |
| Study | Phenotype | Mutation Type | Nature | cDNA | Amino Acid Change | Ethnicity |
|---|---|---|---|---|---|---|
| Yao et al., 2022 [32] | PAPA | Splice site | HET | c.-1-2A>T | − | Chinese |
| Yao et al., 2022 [32] | PAPA | Splice site | HET | c.2247-2A>G | − | Chinese |
| Bilal and Ahmad, 2021 [8] | PAPA | Frameshift | HZ | c.143delG | p. 28Cys>Ser48fs* | Pakistani |
| Hayat el at., 2020 [7] | PAPA | Missense | HZ | c.50T>C | p. 17Leu>Ser | Pakistani |
| Ullah et al., 2019 [31] | PAPA/B | Frameshift | HZ | c.591dupA | p. 21Gln>Thr198fs* | Pakistani |
| Ullah et al., 2019 [31] | PAPA/B | Nonsense | HZ | c. 2173A>T | p. Lys 725* | Pakistani |
| Present study | PAPA with cutaneous syndactyly | Missesnse | HZ | c. 3572 C >T | p.1191Pro >Leu | Pakistani |
| Study | Subject | Phenotypes | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PAPA | PAPB | Syndactyly | Camptodactyly | Clinodactyly | |||||||
| Hands | Feet | Hands | Feet | Hands | Feet | Hands | Feet | Hands | Feet | ||
| Present study | IV-2 | ++ | ++ | − | − | − | ++ | − | − | ++ | − |
| IV-3 | + | − | − | − | − | − | − | − | − | − | |
| Yao et al., 2022 [32] | Fetus II-2 | ++ | ++ | − | − | − | − | − | − | − | − |
| Bilal, 2021 [8] | IV-2 | ++ | ++ | − | − | − | ++ | − | − | − | − |
| IV-3 | + | ++ | − | − | − | − | − | − | − | − | |
| Hayat et at., 2020 [7] | IV-1 | ++ | ++ | − | − | − | − | − | ++ | − | − |
| Ullah et al., 2019 [31] | V-1 | ++ | ++ | − | − | − | − | − | − | ++ | − |
| V-2 | + | ++ | + | − | − | − | − | − | ++ | − | |
| Study | Phenotype | Mutation Type | Nature | cDNA | Amino Acid Change | Ethnicity |
|---|---|---|---|---|---|---|
| Bakar et al., 2022 [33] | PAPA/B | Missense | HET | c.1133C>T | p.378Ser>Leu | Pakistani |
| Yousaf et al., 2020 [34] | PAPA/B | Missense | HET | c.1064C>A | p.355Thr>Asn | Pakistani |
| Ullah et al., 2019 [35] | PPD | Missense | HZ | c.1517T>A | p.506Leu>Gln | Pakistani |
| Palencia et al., 2017 [30] | PAPA | Nonsense | HZ | c.337C>T | p.Arg113* | Pakistani |
| Palencia et al., 2017 [30] | Ranging from simple PAP to EVC syndrome | Nonsense | HZ | c.2340G>A | p.Trp780* | Turkish |
| Palencia et al., 2017 [30] | Ranging from simple PAP to EVC syndrome | Nonsense | HZ | c.1930C>T | p.Gln644* | Turkish |
| Present study | PAPA | Nonsense | HZ | c.337C>T | p.Arg113* | Pakistani |
| Study | Member | Phenotypes | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PPD | PAPA | PAPB | Syndactyly | Camptodactyly | Clinodactyly | ||||||||
| Hands | Feet | Hands | Feet | Hands | Feet | Hands | Feet | Hands | Feet | Hands | Feet | ||
| Present study | IV-2 | − | − | + | ++ | − | − | − | + | − | − | + | − |
| IV-5 | − | − | ++ | − | − | − | − | − | − | − | − | − | |
| Bakar et al., 2022 [33] | III-2 | − | − | ++ | ++ | − | − | − | − | − | − | − | − |
| III-9 | − | − | − | − | ++ | − | − | − | − | − | − | − | |
| IV-3 | − | − | ++ | ++ | − | − | − | − | − | − | − | − | |
| IV-4 | − | − | ++ | ++ | − | − | − | − | − | − | − | − | |
| IV-14 | − | − | − | ++ | − | − | − | − | − | − | − | − | |
| Yousaf et al., 2020 [34] | III-4 | − | − | + | − | − | − | − | − | − | − | − | − |
| IV-2 | − | − | − | ++ | − | − | − | ++ | − | − | − | − | |
| IV-3 | − | − | − | − | + | − | − | − | − | − | − | − | |
| IV-6 | − | − | + | + | − | − | − | − | − | − | − | − | |
| IV-8 | − | − | − | − | + | − | − | + | − | − | − | − | |
| Ullah et al., 2019 [35] | VI-4 | + | − | − | − | − | − | − | − | − | − | − | − |
| VI-5 | ++ | − | − | − | − | − | − | − | − | − | − | − | |
| Palencia et al., 2017 [30] | Patient 1With EVC | − | − | ++ | ++ | − | − | − | − | − | − | − | − |
| Patient 2 | − | − | ++ | − | − | − | − | − | − | − | − | − | |
| Patient 3 | − | − | ++ | − | − | − | − | − | − | − | − | − | |
| Patient 4 | No confirmed phenotype | ||||||||||||
| Patient 5 With EVC | − | − | ++ | ++ | − | − | − | − | − | − | − | − | |
| Patient 6 With EVC | − | − | ++ | ++ | − | − | − | − | − | − | − | − | |
| Patient 7 | − | − | + | ++ | − | − | − | − | − | − | − | − | |
| Patient 8 | − | − | − | ++ | − | − | − | − | − | − | − | − | |
| Patient 9 | − | − | − | ++ | − | − | − | − | − | − | − | − | |
| Patient 10 | − | − | − | ++ | − | − | − | − | − | − | − | − | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, S.; Ali, M.Z.; Muzammal, M.; Khan, A.U.; Ikram, M.; Muurinen, M.; Hussain, S.; Loid, P.; Khan, M.A.; Mäkitie, O. Identification of GLI1 and KIAA0825 Variants in Two Families with Postaxial Polydactyly. Genes 2023, 14, 869. https://doi.org/10.3390/genes14040869
Ahmad S, Ali MZ, Muzammal M, Khan AU, Ikram M, Muurinen M, Hussain S, Loid P, Khan MA, Mäkitie O. Identification of GLI1 and KIAA0825 Variants in Two Families with Postaxial Polydactyly. Genes. 2023; 14(4):869. https://doi.org/10.3390/genes14040869
Chicago/Turabian StyleAhmad, Safeer, Muhammad Zeeshan Ali, Muhammad Muzammal, Amjad Ullah Khan, Muhammad Ikram, Mari Muurinen, Shabir Hussain, Petra Loid, Muzammil Ahmad Khan, and Outi Mäkitie. 2023. "Identification of GLI1 and KIAA0825 Variants in Two Families with Postaxial Polydactyly" Genes 14, no. 4: 869. https://doi.org/10.3390/genes14040869