
T-Cell Receptor Repertoire Characteristics Associated with Prognostic Significance in High-Grade Serous Ovarian Carcinoma

Abstract
:1. Introduction
2. Materials and Methods
2.1. RNA Sequencing Data of HGSOC Patients
2.2. TCR Analysis
2.3. RNA Expression Analysis
2.4. TIL Analysis
2.5. Survival Analysis
2.6. Statistical Analysis
3. Results
3.1. Clinical Characteristics of Patients with HGSOC
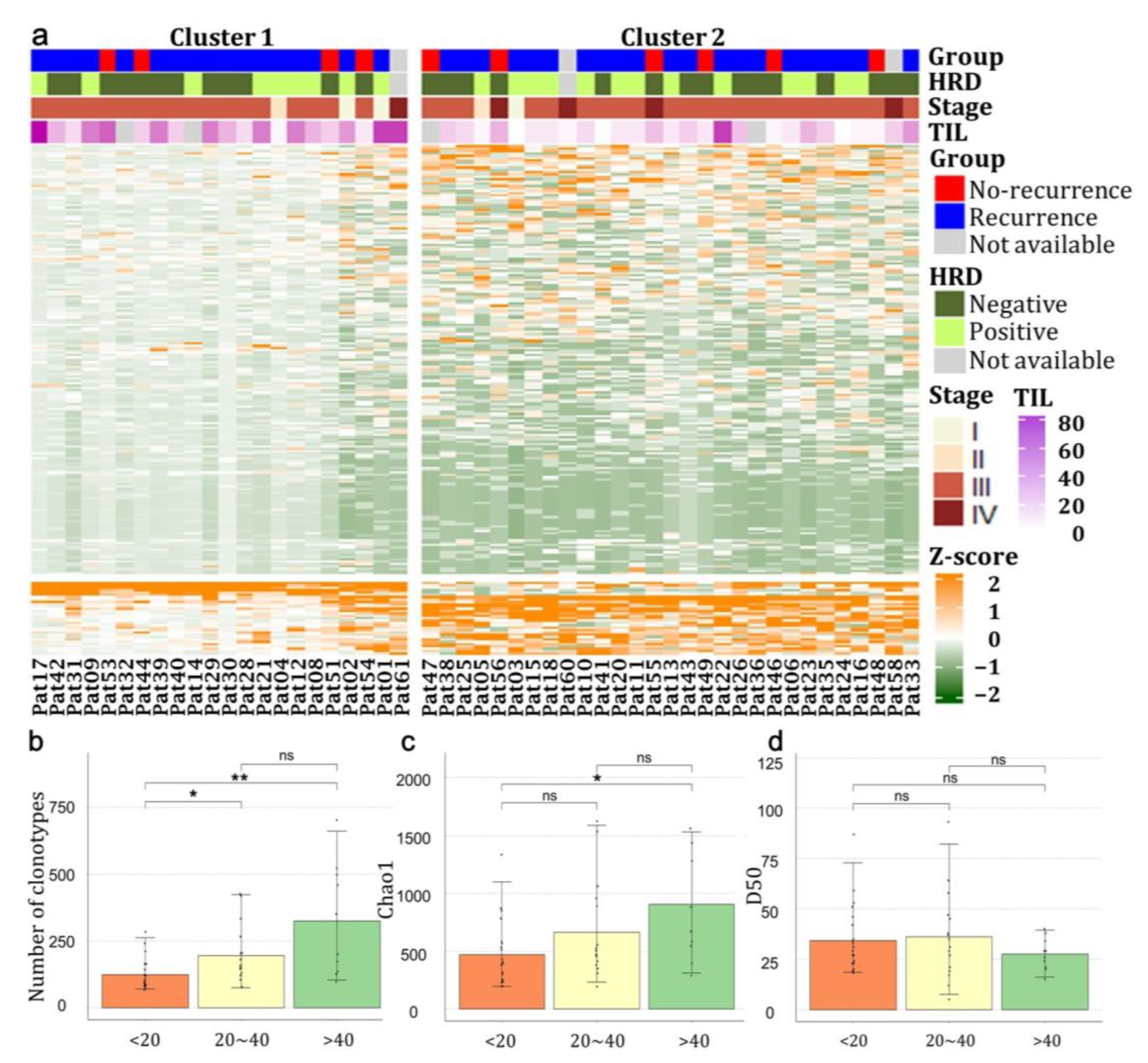
3.2. The Expression Pattern of TCR Repertoire Genes in Patients with HGSOCs
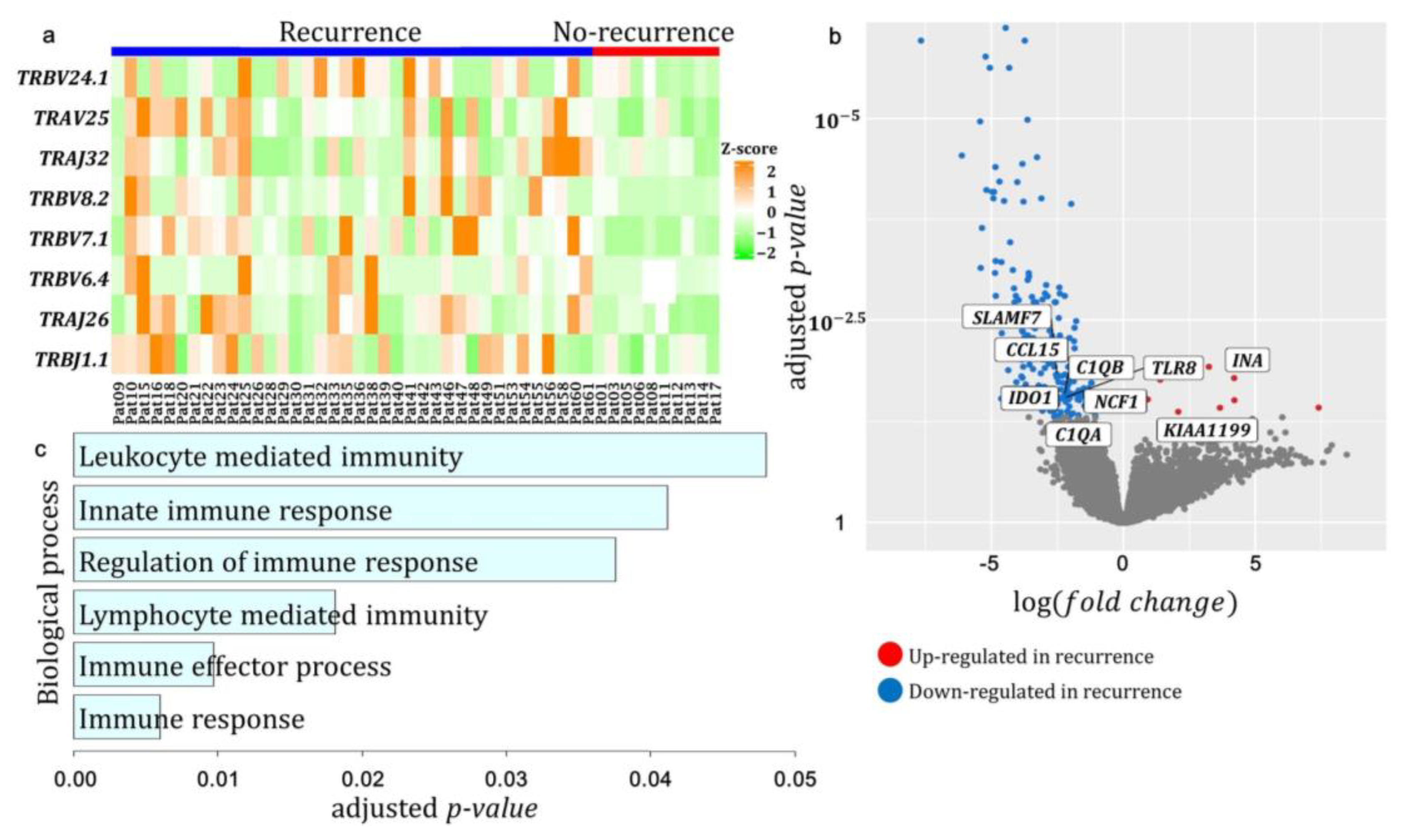
3.3. The Expression Pattern of TCR Repertoire Genes between Recurrence and No-Recurrence Groups
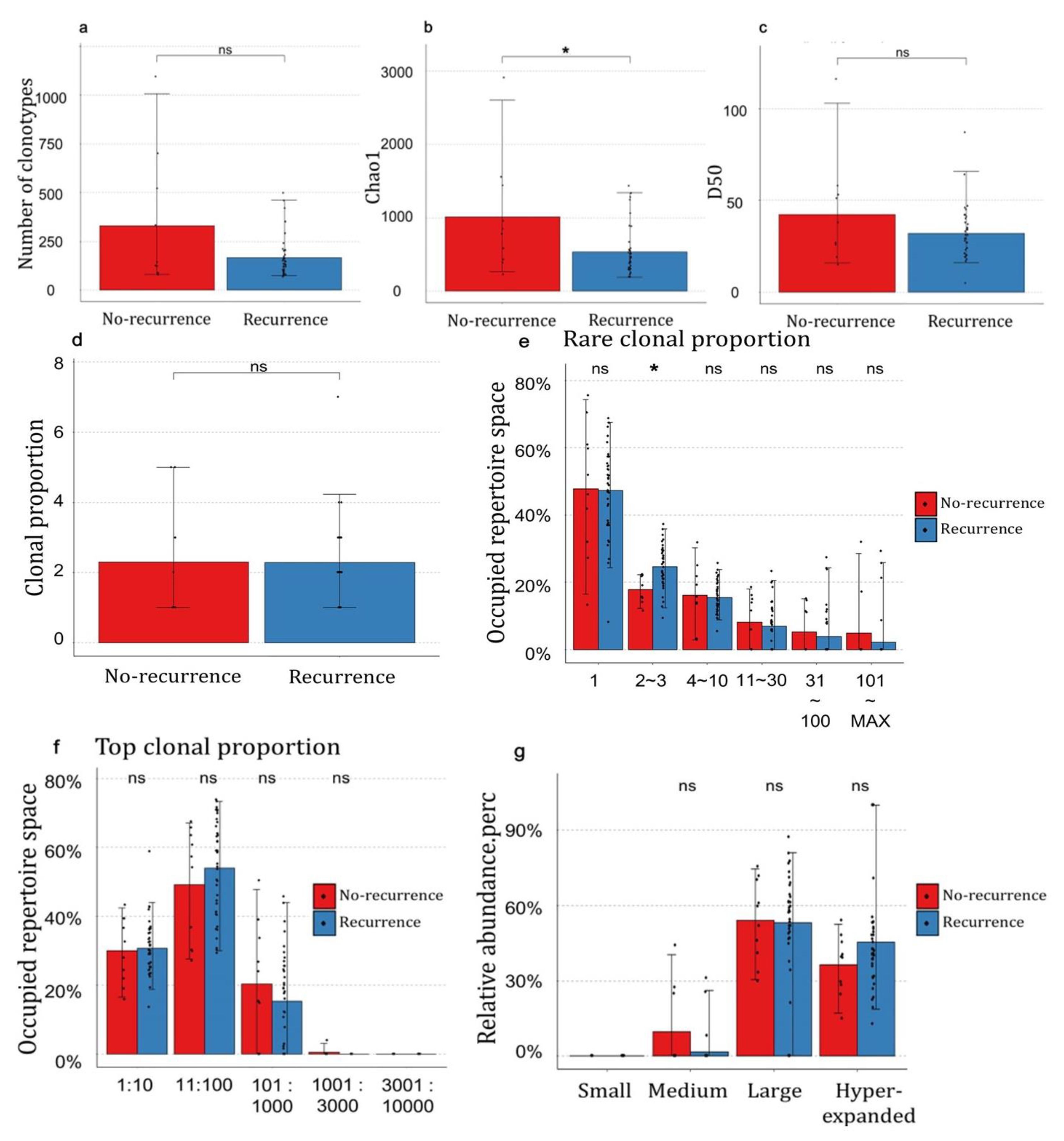
3.4. The Distribution of Clonotypes Occupying TCR Repertoires between Recurrence and No-Recurrence Groups
3.5. Differentially Expressed T-Cell Receptor Gene Segments and Genes between Recurrence and No-Recurrence Groups
3.6. Prognosis of Differentially Expressed Genes in Patients with HGSC
3.7. Differential Expression of TCR Repertoires between HRD Mutation-Positive and Negative Groups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisio, M.A.; Fu, L.; Goyeneche, A.; Gao, Z.H.; Telleria, C. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughan, S.; Coward, J.I.; Bast, R.C.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking Ovarian Cancer: Recommendations for Improving Outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [Green Version]
- Stratton, J.F.; Pharoah, P.; Smith, S.K.; Easton, D.; Ponder, B.A. A Systematic Review and Meta-analysis of Family History and Risk of Ovarian Cancer. Br. J. Obstet. Gynaecol. 1998, 105, 493–499. [Google Scholar] [CrossRef]
- Webb, P.M.; Jordan, S.J. Epidemiology of Epithelial Ovarian Cancer. Best Pract. Res. Clin. Obstet. Gynaecol. 2017, 41, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Integrated Genomic Analyses of Ovarian Carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [Green Version]
- Takaya, H.; Nakai, H.; Takamatsu, S.; Mandai, M.; Matsumura, N. Homologous Recombination Deficiency Status-Based Classification of High-Grade Serous Ovarian Carcinoma. Sci. Rep. 2020, 10, 2757. [Google Scholar] [CrossRef] [Green Version]
- Rosati, E.; Dowds, C.M.; Liaskou, E.; Henriksen, E.K.K.; Karlsen, T.H.; Franke, A. Overview of Methodologies for T-Cell Receptor Repertoire Analysis. BMC Biotechnol. 2017, 17, 61. [Google Scholar] [CrossRef]
- Miles, J.J.; Douek, D.C.; Price, D.A. Bias in the αβ T-Cell Repertoire: Implications for Disease Pathogenesis and Vaccination. Immunol. Cell Biol. 2011, 89, 375–387. [Google Scholar] [CrossRef]
- Kidman, J.; Principe, N.; Watson, M.; Lassmann, T.; Holt, R.A.; Nowak, A.K.; Lesterhuis, W.J.; Lake, R.A.; Chee, J. Characteristics of TCR Repertoire Associated with Successful Immune Checkpoint Therapy Responses. Front. Immunol. 2020, 11, 587014. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.J.; Doherty, P.C.; McCluskey, J.; Rossjohn, J. Structural Determinants of T-Cell Receptor Bias in Immunity. Nat. Rev. Immunol. 2006, 6, 883–894. [Google Scholar] [CrossRef]
- Hong, S.N.; Park, J.Y.; Yang, S.Y.; Lee, C.; Kim, Y.H.; Joung, J.G. Reduced Diversity of Intestinal T-Cell Receptor Repertoire in Patients with Crohn’s Disease. Front. Cell. Infect. Microbiol. 2022, 12, 932373. [Google Scholar] [CrossRef]
- Salgado, R.; Denkert, C.; Demaria, S.; Sirtaine, N.; Klauschen, F.; Pruneri, G.; Wienert, S.; Van den Eynden, G.; Baehner, F.L.; Penault-Llorca, F.; et al. The Evaluation of Tumor-Infiltrating Lymphocytes (TILs) in Breast Cancer: Recommendations by an International TILs Working Group 2014. Ann. Oncol. 2015, 26, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-Infiltrating Lymphocytes in the Immunotherapy Era. Cell. Mol. Immunol. 2021, 18, 842–859. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoiemma, P.P.; Powell, D.J., Jr. Tumor Infiltrating Lymphocytes in Ovarian Cancer. Cancer Biol. Ther. 2015, 16, 807–820. [Google Scholar] [CrossRef]
- Hwang, C.; Lee, S.J.; Lee, J.H.; Kim, K.H.; Suh, D.S.; Kwon, B.S.; Choi, K.U. Stromal Tumor-Infiltrating Lymphocytes Evaluated on H&E-Stained Slides Are an Independent Prognostic Factor in Epithelial Ovarian Cancer and Ovarian Serous Carcinoma. Oncol. Lett. 2019, 17, 4557–4565. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, N.; Wesemann, D.R. Analyzing Immunoglobulin Repertoires. Front. Immunol. 2018, 9, 462. [Google Scholar] [CrossRef] [Green Version]
- Bolotin, D.A.; Poslavsky, S.; Mitrophanov, I.; Shugay, M.; Mamedov, I.Z.; Putintseva, E.V.; Chudakov, D.M. MiXCR: Software for Comprehensive Adaptive Immunity Profiling. Nat. Methods 2015, 12, 380–381. [Google Scholar] [CrossRef]
- Shugay, M.; Bagaev, D.V.; Turchaninova, M.A.; Bolotin, D.A.; Britanova, O.V.; Putintseva, E.V.; Pogorelyy, M.V.; Nazarov, V.I.; Zvyagin, I.V.; Kirgizova, V.I.; et al. VDJtools: Unifying Post-analysis of T Cell Receptor Repertoires. PLoS Comput. Biol. 2015, 11, e1004503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential Expression Analysis of Multifactor RNA-Seq Experiments with Respect to Biological Variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [Green Version]
- Hendry, S.; Salgado, R.; Gevaert, T.; Russell, P.A.; John, T.; Thapa, B.; Christie, M.; van de Vijver, K.; Estrada, M.V.; Gonzalez-Ericsson, P.I.; et al. Assessing Tumor-Infiltrating Lymphocytes in Solid Tumors: A Practical Review for Pathologists and Proposal for a Standardized Method from the International Immuno-Oncology Biomarkers Working Group: Part 2: TILs in Melanoma, Gastrointestinal Tract Carcinomas, Non-small Cell Lung Carcinoma and Mesothelioma, Endometrial and Ovarian Carcinomas, Squamous Cell Carcinoma of the Head and Neck, Genitourinary Carcinomas, and Primary Brain Tumors. Adv. Anat. Pathol. 2017, 24, 311–335. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Choi, M.C.; Kim, S.; Jeong, J.Y.; Kwon, A.Y.; Kim, T.H.; Kim, G.; Joo, W.D.; Park, H.; Lee, C.; et al. USP19 and RPL23 as Candidate Prognostic Markers for Advanced-Stage High-Grade Serous Ovarian Carcinoma. Cancers 2021, 13, 3976. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.C.; Hwang, S.; Kim, S.; Jung, S.G.; Park, H.; Joo, W.D.; Song, S.H.; Lee, C.; Kim, T.H.; Kang, H.; et al. Clinical Impact of Somatic Variants in Homologous Recombination Repair-Related Genes in Ovarian High-Grade Serous Carcinoma. Cancer Res. Treat. 2020, 52, 634–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecuelle, J.; Boidot, R.; Mananet, H.; Derangère, V.; Albuisson, J.; Goussot, V.; Arnould, L.; Tharin, Z.; Ray Coquard, I.; Ghiringhelli, F.; et al. TCR Clonality and Genomic Instability Signatures as Prognostic Biomarkers in High Grade Serous Ovarian Cancer. Cancers 2021, 13, 4394. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, J.N.; Porter, H.; Köbel, M.; Nelson, B.H.; Prentice, L.M.; Kalloger, S.E.; Senz, J.; Milne, K.; Ding, J.; Shah, S.P.; et al. BRCA1 and BRCA2 Mutations Correlate with TP53 Abnormalities and Presence of Immune Cell Infiltrates in Ovarian High-Grade Serous Carcinoma. Mod. Pathol. 2012, 25, 740–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valpione, S.; Mundra, P.A.; Galvani, E.; Campana, L.G.; Lorigan, P.; De Rosa, F.; Gupta, A.; Weightman, J.; Mills, S.; Dhomen, N.; et al. The T Cell Receptor Repertoire of Tumor Infiltrating T Cells Is Predictive and Prognostic for Cancer Survival. Nat. Commun. 2021, 12, 4098. [Google Scholar] [CrossRef]
- Kandalaft, L.E.; Dangaj Laniti, D.; Coukos, G. Immunobiology of High-Grade Serous Ovarian Cancer: Lessons for Clinical Translation. Nat. Rev. Cancer 2022, 22, 640–656. [Google Scholar] [CrossRef]
- Liu, J.; Yan, W.; Han, P.; Tian, D. The Emerging Role of KIAA1199 in Cancer Development and Therapy. Biomed. Pharmacother. 2021, 138, 111507. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Zong, Z.H.; Liu, Y.; Chen, S.; Sheng, X.J.; Zhao, Y. CEMIP Promotes Ovarian Cancer Development and Progression via the PI3K/AKT Signaling Pathway. Biomed. Pharmacother. 2019, 114, 108787. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2019, 51, 411–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Characteristic | Overall (n = 51) | % |
|---|---|---|
| Age (Years), Median (Range) | 55 (36–77) | |
| Stage | ||
| I | 3 | 5.9 |
| II | 2 | 3.9 |
| III | 41 | 80.4 |
| IV | 5 | 9.8 |
| Platinum sensitivity | ||
| No-recurrence | 10 | 19.6 |
| Recurrence | ||
| Platinum sensitive | 26 | 51.0 |
| Platinum resistance | 12 | 23.5 |
| Lost to follow up | 3 | 5.9 |
| TIL % score | ||
| <20 | 21 | 41.2 |
| 20–40 | 16 | 31.4 |
| >40 | 10 | 19.6 |
| Not available | 4 | 7.8 |
| HRD mutation | ||
| Positive | ||
| BRCA1/2 | 18 | 35.3 |
| Others | 4 | 7.8 |
| Negative | 27 | 52.9 |
| Not available | 2 | 3.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-W.; Kim, S.; Yang, S.-Y.; Joung, J.-G.; Hwang, S. T-Cell Receptor Repertoire Characteristics Associated with Prognostic Significance in High-Grade Serous Ovarian Carcinoma. Genes 2023, 14, 785. https://doi.org/10.3390/genes14040785
Kim J-W, Kim S, Yang S-Y, Joung J-G, Hwang S. T-Cell Receptor Repertoire Characteristics Associated with Prognostic Significance in High-Grade Serous Ovarian Carcinoma. Genes. 2023; 14(4):785. https://doi.org/10.3390/genes14040785
Chicago/Turabian StyleKim, Ju-Won, Sewha Kim, So-Yun Yang, Je-Gun Joung, and Sohyun Hwang. 2023. "T-Cell Receptor Repertoire Characteristics Associated with Prognostic Significance in High-Grade Serous Ovarian Carcinoma" Genes 14, no. 4: 785. https://doi.org/10.3390/genes14040785