
Codon Usage Analyses Reveal the Evolutionary Patterns among Plastid Genes of Saxifragales at a Larger-Sampling Scale

Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Retrieval, Reannotation, Alignment, and Compositional Analysis
2.2. The Calculations of Codon Usage Indices
2.3. Graphic Analyses
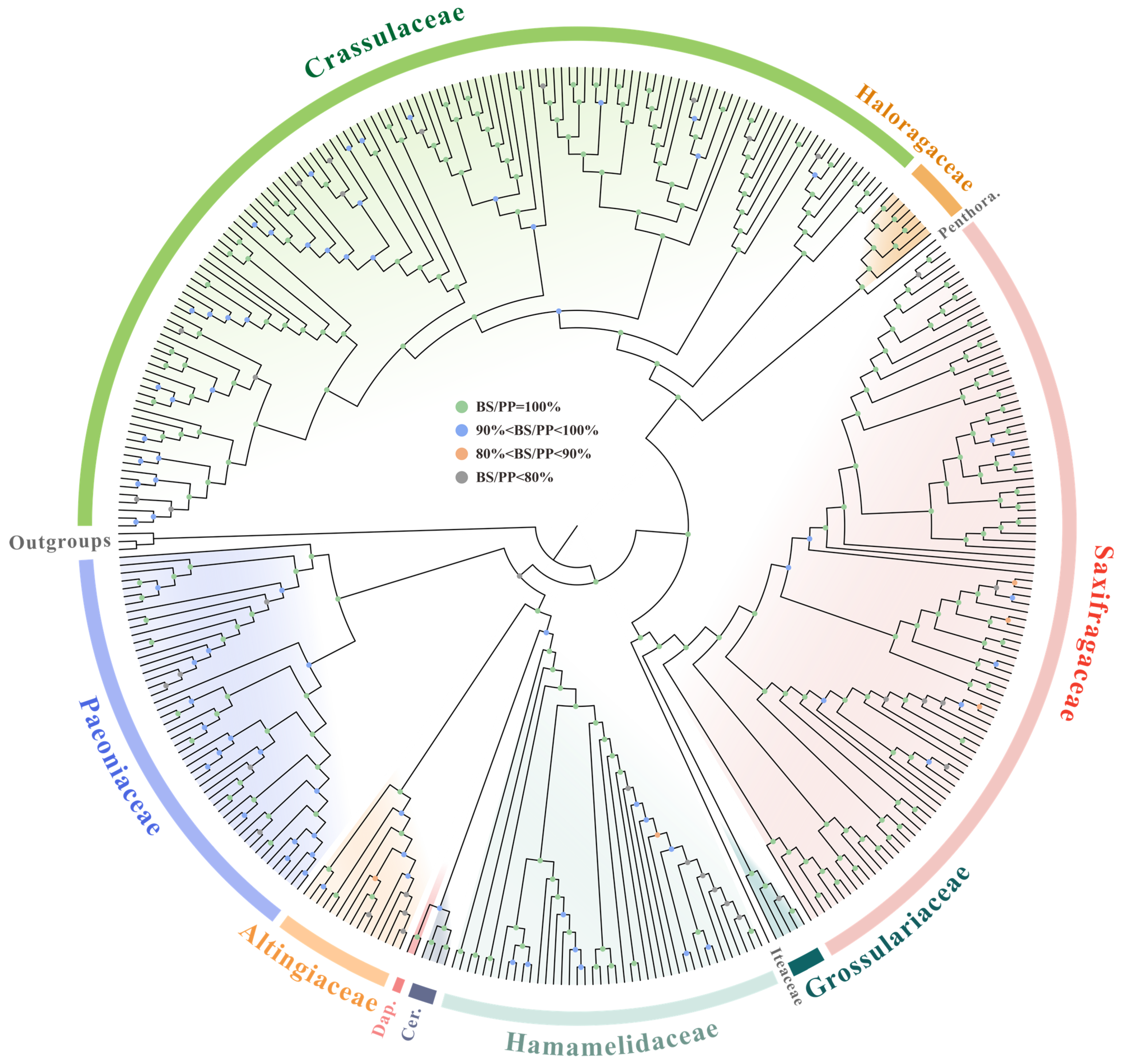
2.4. Phylogenetic Inferences of Saxifragales
3. Results
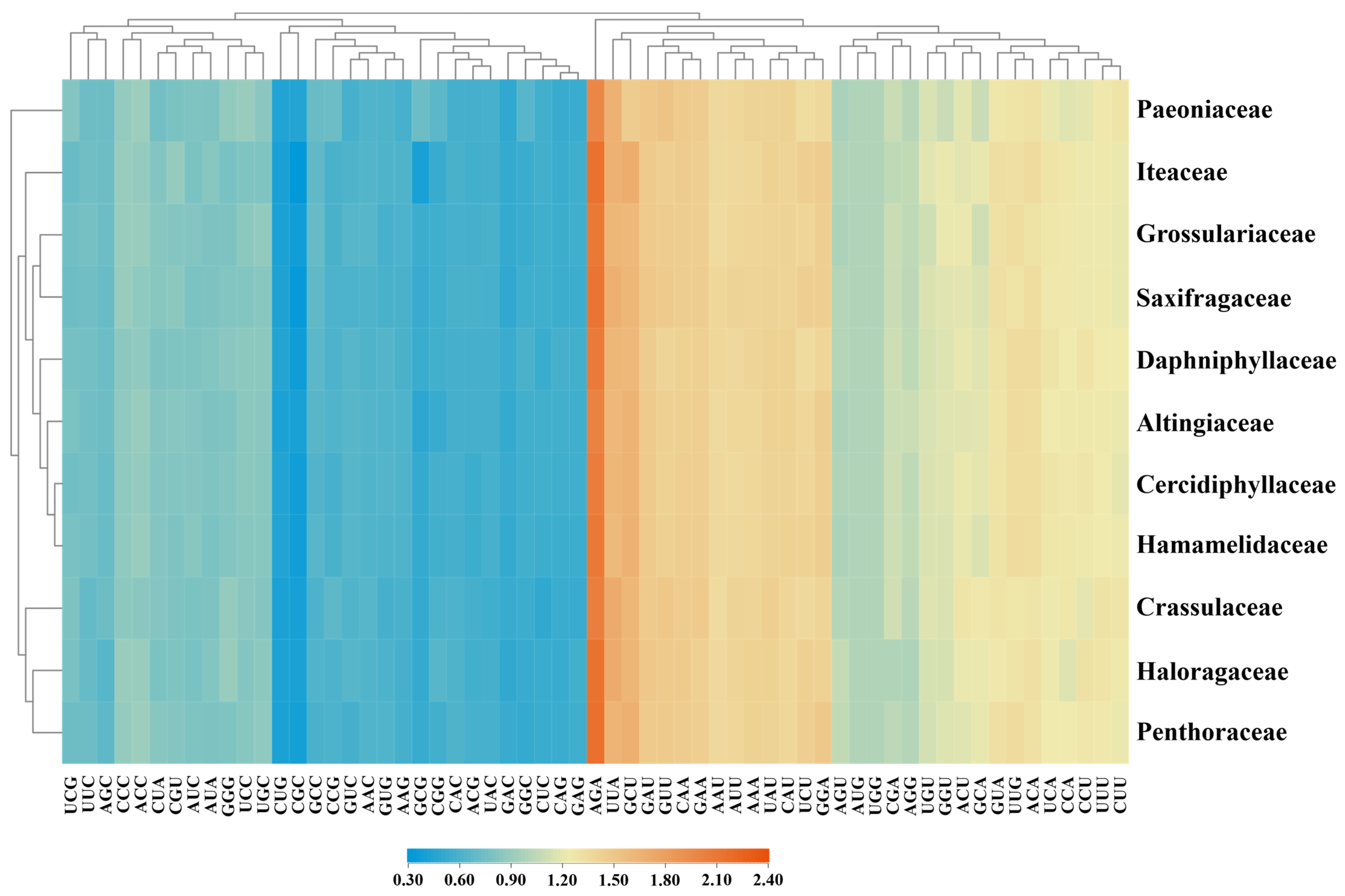
3.1. Base Composition Comparisons of Plastid Genes among Saxifragales
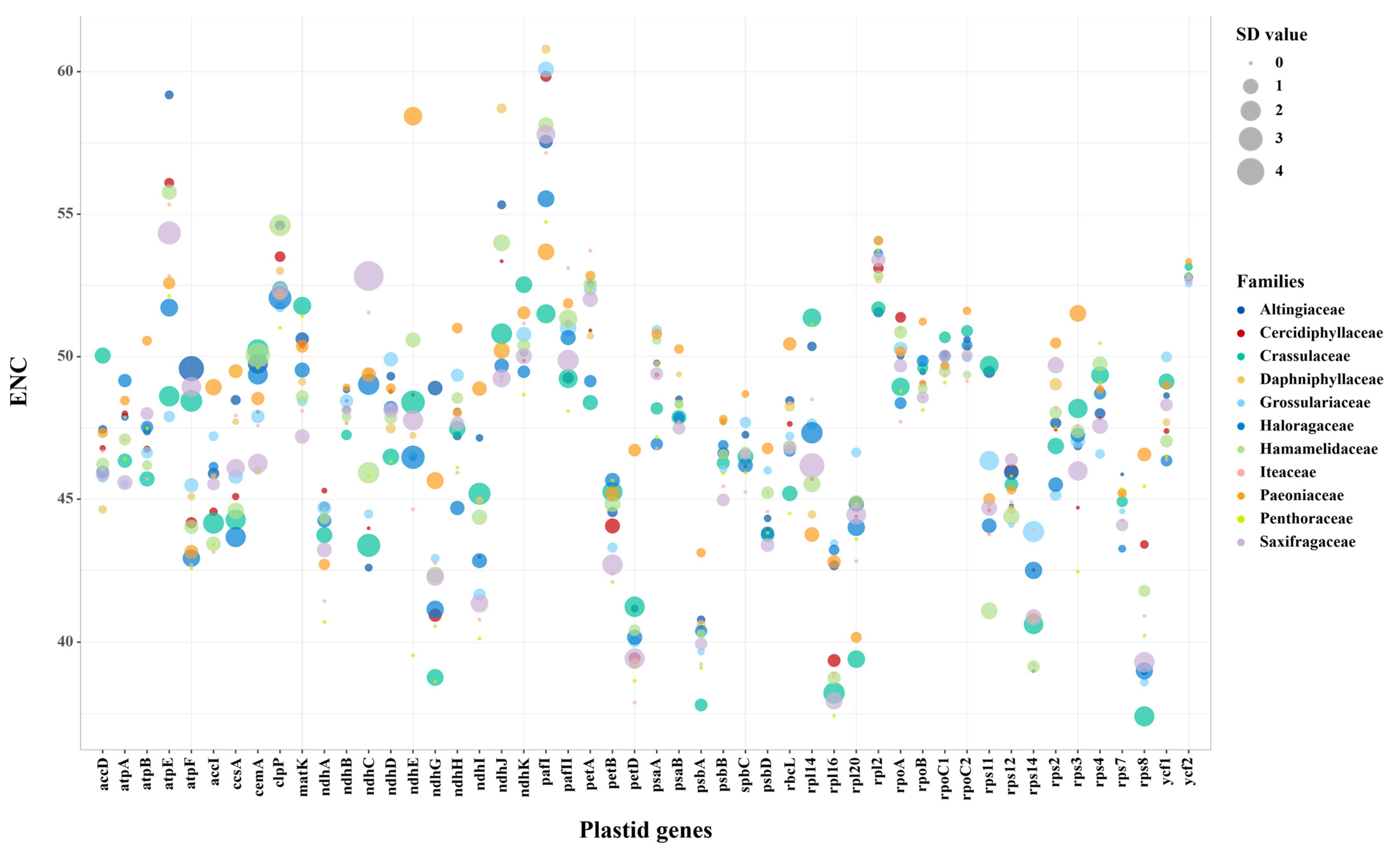
3.2. Codon Usage Bias Indices of Plastid Genes among Saxifragales
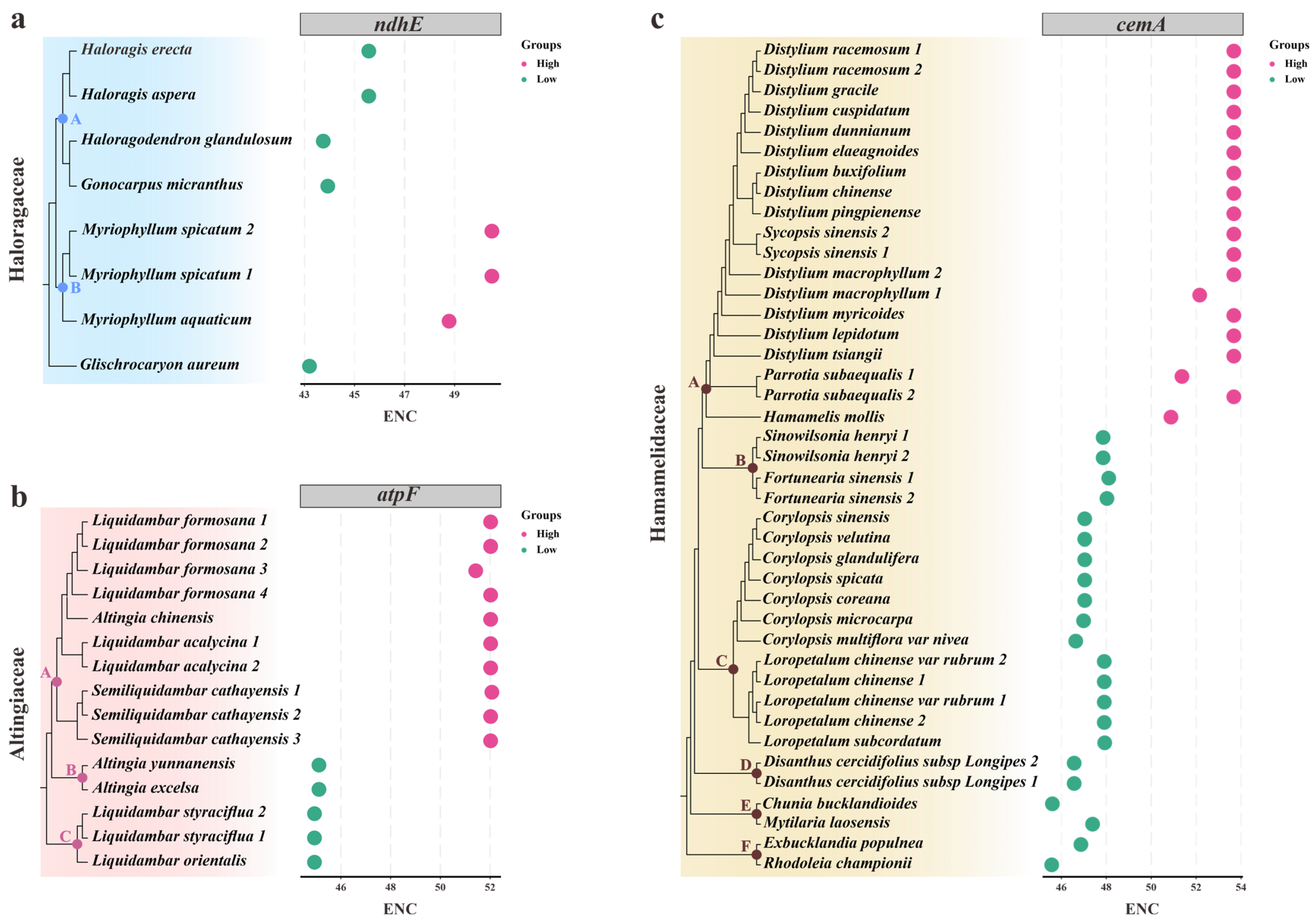
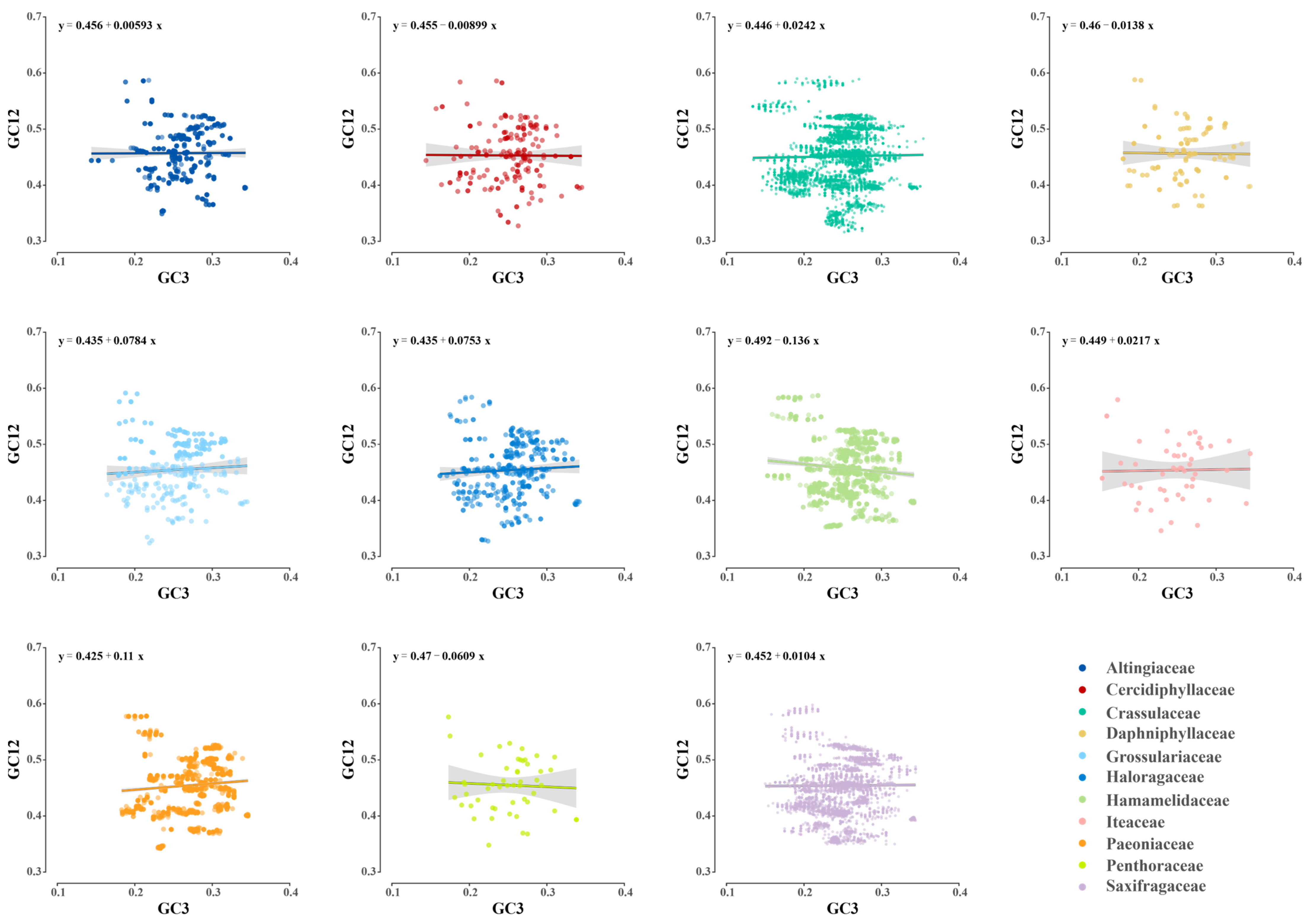
3.3. Determinants in Codon Usage of Plastid Genes among Saxifragales
3.4. Phylogenetic Inferences of Saxifragales Based on Plastid Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jian, S.; Soltis, P.S.; Gitzendanner, M.A.; Moore, M.J.; Li, R.; Hendry, T.A.; Qiu, Y.L.; Dhingra, A.; Bell, C.D.; Soltis, D.E. Resolving an ancient, rapid radiation in Saxifragales. Syst. Biol. 2008, 57, 38–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magallon, S.; Crane, P.R.; Herendeen, P.S. Phylogenetic pattern, diversity, and diversification of eudicots. Ann. Mo. Bot. Gard. 1999, 86, 297–372. [Google Scholar] [CrossRef]
- Moore, M.J.; Soltis, P.S.; Bell, C.D.; Burleigh, J.G.; Soltis, D.E. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. USA 2010, 107, 4623–4628. [Google Scholar] [CrossRef] [Green Version]
- Herna´ ndez-Castillo, G.R.; Cevallos-Ferriz, S.R. Reproductive and vegetative organs with affinities to Haloragaceae from the Upper Cretaceous Huepac Chert Locality of Sonora, Mexico. Am. J. Bot. 1999, 86, 1717–1734. [Google Scholar] [CrossRef]
- Chase, M.W.; Christenhusz, M.J.; Fay, M.F.; Byng, J.; Judd, W.; Soltis, D.; Mabberley, D.; Sennikov, A.; Soltis, P. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Soltis, D.; Soltis, P.; Endress, P.; Chase, M.; Manchester, S.; Judd, W.; Majure, L.; Mavrodiev, E. Phylogeny and Evolution of the Angiosperms; University of Chicago Press: Chicago, IL, USA, 2018. [Google Scholar]
- Takhtadzhian, A.L.; Takhtajan, A. Diversity and Classification of Flowering Plants; Columbia University Press: New York, NY, USA, 1997. [Google Scholar]
- Cronquist, A.; Takhtadzhian, A.L. An Integrated System of Classification of Flowering Plants; Columbia University Press: New York, NY, USA, 1981. [Google Scholar]
- Morgan, D.R.; Soltis, D.E. Phylogenetic relationships among members of Saxifragaceae sensu lato based on rbcL sequence data. Ann. Mo. Bot. Gard. 1993, 80, 631–660. [Google Scholar] [CrossRef]
- Soltis, D.E.; Hibsch-Jetter, C.; Soltis, P.S.; Chase, M.W.; Farris, J.S. Molecular phylogenetic relationships among angiosperms: An overview based on rbcL and 18S rDNA sequences. In Evolution and Diversification of Land Plants, 1st ed.; Iwatsuki, K., Raven, P.H., Eds.; Springer Tokyo: Tokyo, Japan, 1997; pp. 157–178. [Google Scholar]
- Soltis, D.E.; Soltis, P.S. Phylogenetic relationships in Saxifragaceae sensu lato: A comparison of topologies based on 18S rDNA and rbcL sequences. Am. J. Bot. 1997, 84, 504–522. [Google Scholar] [CrossRef] [PubMed]
- Soltis, D.E.; Soltis, P.S.; Nickrent, D.L.; Johnson, L.A.; Hahn, W.J.; Hoot, S.B.; Sweere, J.A.; Kuzoff, R.K.; Kron, K.A.; Chase, M.W. Angiosperm phylogeny inferred from 18S ribosomal DNA sequences. Ann. Mo. Bot. Gard. 1997, 84, 1–49. [Google Scholar] [CrossRef]
- Hoot, S.B.; Magallon, S.; Crane, P.R. Phylogeny of basal eudicots based on three molecular data sets: atpB, rbcL, and 18S nuclear ribosomal DNA sequences. Ann. Mo. Bot. Gard. 1999, 86, 1–32. [Google Scholar] [CrossRef]
- Soltis, D.E.; Soltis, P.S.; Chase, M.W.; Mort, M.E.; Albach, D.C.; Zanis, M.; Savolainen, V.; Hahn, W.H.; Hoot, S.B.; Fay, M.F. Angiosperm phylogeny inferred from 18S rDNA, rbcL, and atpB sequences. Bot. J. Linn. Soc. 2000, 133, 381–461. [Google Scholar] [CrossRef]
- Fishbein, M.; Hibsch-Jetter, C.; Soltis, D.E.; Hufford, L. Phylogeny of Saxifragales (angiosperms, eudicots): Analysis of a rapid, ancient radiation. Syst. Biol. 2001, 50, 817–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishbein, M.; Soltis, D.E. Further resolution of the rapid radiation of Saxifragales (angiosperms, eudicots) supported by mixed-model Bayesian analysis. Syst. Bot. 2004, 29, 883–891. [Google Scholar] [CrossRef]
- Soltis, D.E.; Mort, M.E.; Latvis, M.; Mavrodiev, E.V.; O’Meara, B.C.; Soltis, P.S.; Burleigh, J.G.; Rubio de Casas, R. Phylogenetic relationships and character evolution analysis of Saxifragales using a supermatrix approach. Am. J. Bot. 2013, 100, 916–929. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Zhu, R.; Dong, J.; Bi, D.; Jiang, L.; Zeng, J.; Huang, Q.; Liu, H.; Xu, W.; Wu, L.; et al. Next-Generation Genome Sequencing of Sedum. plumbizincicola Sheds Light on the Structural Evolution of Plastid rRNA Operon and Phylogenetic Implications within Saxifragales. Plants 2019, 8, 386. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Bi, D.; Yi, R.; Ding, H.; Wu, L.; Kan, X. Plastome evolution of Aeonium and Monanthes (Crassulaceae): Insights into the variation of plastomic tRNAs, and the patterns of codon usage and aversion. Planta 2022, 256, 35. [Google Scholar] [CrossRef]
- Han, S.; Ding, H.; Bi, D.; Zhang, S.; Yi, R.; Gao, J.; Yang, J.; Ye, Y.; Wu, L.; Kan, X. Structural Diversities and Phylogenetic Signals in Plastomes of the Early-Divergent Angiosperms: A Case Study in Saxifragales. Plants 2022, 11, 3544. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Cheng, T.; Zhou, S. Complete chloroplast genome of Sedum. sarmentosum and chloroplast genome evolution in Saxifragales. PLoS ONE 2013, 8, e77965. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Xu, C.; Wu, P.; Cheng, T.; Yu, J.; Zhou, S.; Hong, D.Y. Resolving the systematic positions of enigmatic taxa: Manipulating the chloroplast genome data of Saxifragales. Mol. Phylogenet Evol. 2018, 126, 321–330. [Google Scholar] [CrossRef]
- Tarullo, C.; Rose, J.; Sytsma, K.; Drew, B.T. Using a supermatrix approach to explore phylogenetic relationships, divergence times, and historical biogeography of Saxifragales. Turk. J. Botany 2021, 45, 5. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Wang, Y.; He, P.; Li, P.; Lee, J.; Soltis, D.E.; Fu, C. Chloroplast genome analyses and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data. BMC Genom. 2018, 19, 235. [Google Scholar] [CrossRef] [Green Version]
- Henriquez, C.L.; Abdullah; Ahmed, I.; Carlsen, M.M.; Zuluaga, A.; Croat, T.B.; McKain, M.R. Molecular evolution of chloroplast genomes in Monsteroideae (Araceae). Planta 2020, 251, 72. [Google Scholar] [CrossRef] [PubMed]
- Shahzadi, I.; Abdullah; Mehmood, F.; Ali, Z.; Ahmed, I.; Mirza, B. Chloroplast genome sequences of Artemisia maritima and Artemisia absinthium: Comparative analyses, mutational hotspots in genus Artemisia and phylogeny in family Asteraceae. Genomics 2020, 112, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.P.; Guo, W.; Partha, R.; Fan, W.; Levsen, N.; Wolff, K.; Nugent, J.M.; Pabon-Mora, N.; Gonzalez, F. Plastomes from tribe Plantagineae (Plantaginaceae) reveal infrageneric structural synapormorphies and localized hypermutation for Plantago and functional loss of ndh genes from Littorella. Mol. Phylogenet. Evol. 2021, 162, 107217. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Yang, Y.; Zhou, T.; Liu, Z.-L. Comparative plastid genomes of Primula species: Sequence divergence and phylogenetic relationships. Int. J. Mol. Sci. 2018, 19, 1050. [Google Scholar] [CrossRef] [Green Version]
- Hong, Z.; Wu, Z.; Zhao, K.; Yang, Z.; Zhang, N.; Guo, J.; Tembrock, L.R.; Xu, D. Comparative analyses of five complete chloroplast genomes from the genus Pterocarpus (Fabacaeae). Int. J. Mol. Sci. 2020, 21, 3758. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Lou, G.; Cai, X.; Zhang, B.; Cheng, Y.; Wang, H. Comparison of the complete plastomes and the phylogenetic analysis of Paulownia species. Sci. Rep. 2020, 10, 2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco, T.G.; Lopes, A.d.S.; Welter, J.F.; Yotoko, K.S.C.; Otoni, W.C.; Vieira, L.d.N.; Guerra, M.P.; Nodari, R.O.; Balsanelli, E.; Pedrosa, F.d.O. Plastome sequences of the subgenus Passiflora reveal highly divergent genes and specific evolutionary features. Plant Mol. Biol. 2020, 104, 21–37. [Google Scholar] [CrossRef]
- Chen, S.; Wang, T.; Shu, J.; Xiang, Q.; Yang, T.; Zhang, X.; Yan, Y. Plastid Phylogenomics and Plastomic Diversity of the Extant Lycophytes. Genes 2022, 13, 1280. [Google Scholar] [CrossRef]
- Sheng, J.; She, X.; Liu, X.; Wang, J.; Hu, Z. Comparative analysis of codon usage patterns in chloroplast genomes of five Miscanthus species and related species. PeerJ 2021, 9, e12173. [Google Scholar] [CrossRef]
- Suzuki, H.; Morton, B.R. Codon Adaptation of Plastid Genes. PLoS ONE 2016, 11, e0154306. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Nie, X.; Jia, X.; Zhao, C.; Biradar, S.S.; Wang, L.; Du, X.; Weining, S. Analysis of codon usage patterns of the chloroplast genomes in the Poaceae family. Aust. J. Bot. 2012, 60, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Ikemura, T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: A proposal for a synonymous codon choice that is optimal for the E. coli translational system. J. Mol. Biol. 1981, 151, 389–409. [Google Scholar] [CrossRef]
- Wang, L.; Roossinck, M.J. Comparative analysis of expressed sequences reveals a conserved pattern of optimal codon usage in plants. Plant. Mol. Biol. 2006, 61, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Vicario, S.; Mason, C.E.; White, K.P.; Powell, J.R. Developmental stage and level of codon usage bias in Drosophila. Mol. Biol. Evol. 2008, 25, 2269–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, H.E.; Lawrie, D.S.; Petrov, D.A. Pervasive strong selection at the level of codon usage bias in Drosophila melanogaster. Genetics 2020, 214, 511–528. [Google Scholar] [CrossRef] [Green Version]
- Iriarte, A.; Lamolle, G.; Musto, H. Codon Usage Bias: An Endless Tale. J. Mol. Evol. 2021, 89, 589–593. [Google Scholar] [CrossRef]
- Yang, J.; Ding, H.; Kan, X. Codon usage patterns and evolution of HSP60 in birds. Int. J. Biol. Macromol. 2021, 183, 1002–1012. [Google Scholar] [CrossRef]
- Ding, H.; Bi, D.; Han, S.; Yi, R.; Zhang, S.; Ye, Y.; Gao, J.; Yang, J.; Kan, X. Mitogenomic Codon Usage Patterns of Superfamily Certhioidea (Aves, Passeriformes): Insights into Asymmetrical Bias and Phylogenetic Implications. Animals 2022, 13, 96. [Google Scholar] [CrossRef]
- Parvathy, S.T.; Udayasuriyan, V.; Bhadana, V. Codon usage bias. Mol. Biol. Rep. 2022, 49, 539–565. [Google Scholar] [CrossRef]
- Salim, H.M.; Cavalcanti, A.R. Factors influencing codon usage bias in genomes. J. Braz. Chem. Soc. 2008, 19, 257–262. [Google Scholar] [CrossRef] [Green Version]
- Grantham, R.; Gautier, C.; Gouy, M.; Mercier, R.; Pave, A. Codon catalog usage and the genome hypothesis. Nucleic. Acids. Res. 1980, 8, 197. [Google Scholar] [CrossRef]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.-H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef]
- Sueoka, N. Intrastrand parity rules of DNA base composition and usage biases of synonymous codons. J. Mol. Evol. 1995, 40, 318–325. [Google Scholar] [CrossRef]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Yu, X.; Yang, Y.; Wei, P.; Zhang, W.; Li, X.; Liu, C.; Zhao, S.; Li, X.; Liu, X. Comparative Analysis of Chloroplast Genomes within Saxifraga (Saxifragaceae) Takes Insights into Their Genomic Evolution and Adaption to the High-Elevation Environment. Genes 2022, 13, 1673. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Han, S.; Ye, Y.; Bi, D.; Zhang, S.; Yi, R.; Gao, J.; Yang, J.; Wu, L.; Kan, X. Ten Plastomes of Crassula (Crassulaceae) and Phylogenetic Implications. Biology 2022, 11, 1779. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids. Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic. Acids. Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Xia, X. DAMBE7: New and improved tools for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McInerney, J.O. Replicational and transcriptional selection on codon usage in Borrelia. burgdorferi. Proc. Natl. Acad. Sci. USA 1998, 95, 10698–10703. [Google Scholar] [CrossRef] [Green Version]
- Roth, A.; Anisimova, M.; Cannarozzi, G.M. Measuring codon usage bias. In Codon Evolution: Mechanisms and Models, 1st ed.; Cannarozzi, G.M., Schneider, A., Eds.; Oxford University Press: Oxford, UK, 2012; pp. 189–217. [Google Scholar]
- Wang, L.; Xing, H.; Yuan, Y.; Wang, X.; Saeed, M.; Tao, J.; Feng, W.; Zhang, G.; Song, X.; Sun, X. Genome-wide analysis of codon usage bias in four sequenced cotton species. PLoS ONE 2018, 13, e0194372. [Google Scholar] [CrossRef] [Green Version]
- Peden, J.F. Analysis of Codon Usage. Ph.D. Thesis, University of Nottingham, Nottingham, UK, 2000. [Google Scholar]
- Li, G.; Zhang, L.; Xue, P. Codon usage pattern and genetic diversity in chloroplast genomes of Panicum species. Gene 2021, 802, 145866. [Google Scholar] [CrossRef] [PubMed]
- Comeron, J.M.; Aguadé, M. An evaluation of measures of synonymous codon usage bias. J. Mol. Evol. 1998, 47, 268–274. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Gan, H.; Liang, X. Analysis of synonymous codon usage bias in potato virus M and its adaption to hosts. Viruses 2019, 11, 752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. Ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Galtier, N.; Lobry, J.R. Relationships between genomic G+ C content, RNA secondary structures, and optimal growth temperature in prokaryotes. J. Mol. Evol. 1997, 44, 632–636. [Google Scholar] [CrossRef]
- Sueoka, N. Translation-coupled violation of Parity Rule 2 in human genes is not the cause of heterogeneity of the DNA G+ C content of third codon position. Gene 1999, 238, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Sueoka, N. Two aspects of DNA base composition: G+ C content and translation-coupled deviation from intra-strand rule of A=T and G=C. J. Mol. Evol. 1999, 49, 49–62. [Google Scholar] [CrossRef]
- Yu, X.; Liu, J.; Li, H.; Liu, B.; Zhao, B.; Ning, Z. Comprehensive analysis of synonymous codon usage patterns and influencing factors of porcine epidemic diarrhea virus. Arch. Virol. 2021, 166, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Zhang, R.; Butler III, R.R.; Liu, T.; Zhang, L.; Pombert, J.-F.; Zhou, Z. Comparative analysis of codon usage bias patterns in microsporidian genomes. PLoS ONE 2015, 10, e0129223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Pan, Z.; Gao, S.; He, Y.; Xia, Q.; Jin, Y.; Yao, H. Analysis of synonymous codon usage of chloroplast genome in Porphyra umbilicalis. Genes Genom. 2019, 41, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Indrabalan, U.B.; Suresh, K.P.; Shivamallu, C.; Patil, S.S. An extensive evaluation of codon usage pattern and bias of structural proteins p30, p54 and, p72 of the African swine fever virus (ASFV). Virusdisease 2021, 32, 810–822. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Plotkin, J.B.; Kudla, G. Synonymous but not the same: The causes and consequences of codon bias. Nat. Rev. Genet. 2011, 12, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Xue, Q. Comparative studies on codon usage pattern of chloroplasts and their host nuclear genes in four plant species. J. Genet. 2005, 84, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Bellgard, M.; Schibeci, D.; Trifonov, E.; Gojobori, T. Early detection of G + C differences in bacterial species inferred from the comparative analysis of the two completely sequenced Helicobacter pylori strains. J. Mol. Evol. 2001, 53, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Clegg, M.T.; Gaut, B.S.; Learn, G.H., Jr.; Morton, B.R. Rates and patterns of chloroplast DNA evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 6795–6801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howe, C.J.; Barbrook, A.C.; Koumandou, V.L.; Nisbet, R.E.R.; Symington, H.A.; Wightman, T.F. Evolution of the chloroplast genome. Philos. Trans. R Soc. Lond. B Biol. Sci. 2003, 358, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.J.; Zhou, J.; Li, Z.F.; Wang, L.; Gu, X.; Zhong, Y. Comparative analysis of codon usage patterns among mitochondrion, chloroplast and nuclear genes in Triticum aestivum L. J. Integr. Plant. Biol. 2007, 49, 246–254. [Google Scholar] [CrossRef]
- Yengkhom, S.; Uddin, A.; Chakraborty, S. Deciphering codon usage patterns and evolutionary forces in chloroplast genes of Camellia sinensis var assamica and Camellia sinensis var sinensis in comparison to Camellia pubicosta. J. Integr. Agric. 2019, 18, 2771–2785. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, B.; Li, B.; Zhou, Q.; Wang, G.; Jiang, X.; Wang, C.; Xu, Z. Comparative analysis of codon usage patterns in chloroplast genomes of six Euphorbiaceae species. PeerJ 2020, 8, e8251. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Cai, Q.; Wang, Y.; Li, M.; Wang, C.; Wang, Z.; Jiao, C.; Xu, C.; Wang, H.; Zhang, Z. Comparative analysis of codon Bias in the chloroplast genomes of Theaceae species. Front. Genet. 2022, 13, 824610. [Google Scholar] [CrossRef]
- Morton, B.R.; So, B.G. Codon usage in plastid genes is correlated with context, position within the gene, and amino acid content. J. Mol. Evol. 2000, 50, 184–193. [Google Scholar] [CrossRef]
- Kandeel, M.; Ibrahim, A.; Fayez, M.; Al-Nazawi, M. From SARS and MERS CoVs to SARS-CoV-2: Moving toward more biased codon usage in viral structural and nonstructural genes. J. Med. Virol. 2020, 92, 660–666. [Google Scholar] [CrossRef] [Green Version]
- Nair, R.R.; Raveendran, N.T.; Dirisala, V.R.; Nandhini, M.B.; Sethuraman, T.; Venkateswarulu, T.C.; Doss, G. Mutational pressure drives evolution of synonymous codon usage in genetically distinct Oenothera plastomes. Iran. J. Biotechnol. 2014, 12, e1156. [Google Scholar] [CrossRef] [Green Version]
- Bulmer, M. Are codon usage patterns in unicellular organisms determined by selection-mutation balance? J. Evol. Biol. 1988, 1, 15–26. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, L.; Wang, W.; Zhang, Z.; Du, H.; Qu, Z.; Li, X.-Q.; Xiang, H. Differences in codon usage bias between photosynthesis-related genes and genetic system-related genes of chloroplast genomes in cultivated and wild solanum species. Int. J. Mol. Sci. 2018, 19, 3142. [Google Scholar] [CrossRef] [Green Version]
- Kawabe, A.; Miyashita, N.T. Patterns of codon usage bias in three dicot and four monocot plant species. Genes Genet. Syst. 2003, 78, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Campbell, W.H.; Gowri, G. Codon usage in higher plants, green algae, and cyanobacteria. Plant. Physiol. 1990, 92, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.; Gan, Z.; Zhang, F.; Yi, X.; Zhang, J.; Wan, X. Analysis of codon usage patterns in citrus based on coding sequence data. BMC Genom. 2020, 21, 234. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Gojobori, T.; Ikemura, T. Codon usage tabulated from international DNA sequence databases: Status for the year 2000. Nucleic Acids Res. 2000, 28, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouy, M.; Gautier, C. Codon usage in bacteria: Correlation with gene expressivity. Nucleic Acids Res. 1982, 10, 7055–7074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulmer, M. The selection-mutation-drift theory of synonymous codon usage. Genetics 1991, 129, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.; Wu, G.; Wang, Z.; Chai, X.; Nie, Q.; Zhang, X. Mutation bias is the driving force of codon usage in the Gallus gallus genome. DNA Res. 2011, 18, 499–512. [Google Scholar] [CrossRef] [Green Version]
- Guan, D.-L.; Ma, L.-B.; Khan, M.S.; Zhang, X.-X.; Xu, S.-Q.; Xie, J.-Y. Analysis of codon usage patterns in Hirudinaria. manillensis reveals a preference for GC-ending codons caused by dominant selection constraints. BMC Genom. 2018, 19, 542. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.K.; Baruah, V.J.; Satapathy, S.S.; Banerjee, R. Cotranslational protein folding reveals the selective use of synonymous codons along the coding sequence of a low expression gene. J. Genet. 2014, 93, 613–617. [Google Scholar] [CrossRef]
- Somaratne, Y.; Guan, D.-L.; Wang, W.-Q.; Zhao, L.; Xu, S.-Q. The complete chloroplast genomes of two Lespedeza species: Insights into codon usage bias, RNA editing sites, and phylogenetic relationships in Desmodieae (Fabaceae: Papilionoideae). Plants 2019, 9, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Wei, F.; Cai, Z.; Wei, Y.; Khan, A.; Miao, J.; Wei, K. Analysis of codon usage bias and evolution in the chloroplast genome of Mesona. chinensis Benth. Dev. Genes Evol. 2021, 231, 1–9. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, D.; Guo, K.; Zhao, L.; Meng, F.; Xiao, J.; Niu, Y.; Sun, Y. Comparative analysis of codon usage patterns in chloroplast genomes of ten Epimedium species. BMC Genom. Data 2023, 24, 3. [Google Scholar] [CrossRef]
- Wang, Y.; Yao, L.; Fan, J.; Zhao, X.; Zhang, Q.; Chen, Y.; Guo, C. The Codon Usage Bias Analysis of Free-Living Ciliates’ Macronuclear Genomes and Clustered Regularly Interspaced Short Palindromic Repeats/Cas9 Vector Construction of Stylonychia lemnae. Front. Microbiol. 2022, 13, 785889. [Google Scholar] [CrossRef]
- Morton, B.R. Context-dependent mutation dynamics, not selection, explains the codon usage bias of most angiosperm chloroplast genes. J. Mol. Evol. 2022, 90, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Zhang, Q.; Wang, C.; Li, F.; Tian, F.; Lu, Y.; Hu, Y.; Yang, H.; Cui, G. Analysis of codon usage patterns of the chloroplast genome in Delphinium grandiflorum L. reveals a preference for AT-ending codons as a result of major selection constraints. PeerJ 2021, 9, e10787. [Google Scholar] [CrossRef]
- Liu, H.; Lu, Y.; Lan, B.; Xu, J. Codon usage by chloroplast gene is bias in Hemiptelea davidii. J. Genet. 2020, 99, 8. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Cai, X.; Chen, Q.; Zhou, H.; Cai, Y.; Ben, A. Factors affecting synonymous codon usage bias in chloroplast genome of oncidium gower ramsey. Evol. Bioinform. 2011, 7, 271–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | GC% | GC1% | GC2% | GC3% |
|---|---|---|---|---|
| Altingiaceae | 39.29 ± 3.31 | 48.47 ± 5.45 | 40.05 ± 5.75 | 29.35 ± 3.21 |
| Cercidiphyllaceae | 38.67 ± 3.44 | 48.05 ± 5.48 | 39.65 ± 5.83 | 28.31 ± 3.59 |
| Crassulaceae | 38.42 ± 3.53 | 47.84 ± 5.56 | 39.54 ± 5.76 | 27.89 ± 3.69 |
| Daphniphyllaceae | 39.20 ± 3.25 | 48.43 ± 5.38 | 40.01 ± 5.57 | 29.15 ± 3.53 |
| Grossulariaceae | 38.83 ± 3.44 | 48.18 ± 5.52 | 39.82 ± 5.58 | 28.49 ± 3.74 |
| Haloragaceae | 38.79 ± 3.45 | 48.15 ± 5.51 | 39.78 ± 5.61 | 28.45 ± 3.57 |
| Hamamelidaceae | 39.15 ± 3.19 | 48.36 ± 5.51 | 40.11 ± 5.64 | 28.97 ± 3.34 |
| Iteaceae | 38.59 ± 3.59 | 48.04 ± 5.76 | 39.87 ± 5.83 | 27.87 ± 3.96 |
| Paeoniaceae | 39.46 ± 3.47 | 48.26 ± 5.34 | 39.86 ± 5.55 | 30.27 ± 3.60 |
| Penthoraceae | 38.75 ± 3.30 | 48.26 ± 5.56 | 39.88 ± 5.51 | 28.12 ± 3.57 |
| Saxifragaceae | 38.77 ± 3.43 | 48.23 ± 5.44 | 39.76 ± 5.77 | 28.33 ± 3.60 |
| Gene | ENC Values | Gene | ENC Values | Gene | ENC Values | Gene | ENC Values | Gene | ENC Values |
|---|---|---|---|---|---|---|---|---|---|
| rpl16 | 39.23 ± 2.50 | ndhI | 44.48 ± 3.03 | atpF | 47.13 ± 3.09 | rps2 | 48.28 ± 1.81 | rpoC1 | 50.16 ± 0.63 |
| psbA | 39.67 ± 1.82 | rps7 | 44.77 ± 0.67 | atpB | 47.21 ± 1.78 | ycf1 | 48.53 ± 1.09 | rpoC2 | 50.54 ± 0.77 |
| rps8 | 39.92 ± 3.46 | atpI | 45.28 ± 2.24 | ndhC | 47.27 ± 5.15 | rps4 | 48.72 ± 1.40 | petA | 50.78 ± 2.10 |
| rps14 | 40.60 ± 1.56 | rps12 | 45.56 ± 0.95 | ndhD | 47.66 ± 1.36 | cemA | 48.76 ± 2.68 | ndhJ | 50.86 ± 2.45 |
| petD | 41.32 ± 2.81 | ccsA | 45.77 ± 2.42 | accD | 47.74 ± 2.03 | psaA | 49.24 ± 1.19 | ndhK | 51.21 ± 1.53 |
| ndhG | 41.66 ± 3.06 | psbB | 46.21 ± 1.06 | rps3 | 47.80 ± 2.36 | rpoB | 49.45 ± 0.93 | atpE | 52.19 ± 3.73 |
| rpl20 | 42.05 ± 2.88 | rps11 | 46.40 ± 3.25 | ndhB | 47.92 ± 0.67 | rpoA | 49.62 ± 1.27 | clpP | 52.68 ± 1.53 |
| ndhA | 43.59 ± 1.06 | atpA | 46.66 ± 1.27 | rpl14 | 47.94 ± 3.61 | ndhE | 49.68 ± 4.29 | rpl2 | 52.72 ± 1.15 |
| psbD | 44.30 ± 1.33 | rbcL | 46.80 ± 1.89 | ndhH | 48.09 ± 1.57 | matK | 49.78 ± 2.19 | ycf2 | 52.98 ± 0.28 |
| petB | 44.42 ± 2.04 | psbC | 46.87 ± 1.06 | psaB | 48.20 ± 1.09 | pafII | 50.11 ± 1.95 | pafI | 54.91 ± 3.38 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bi, D.; Han, S.; Zhou, J.; Zhao, M.; Zhang, S.; Kan, X. Codon Usage Analyses Reveal the Evolutionary Patterns among Plastid Genes of Saxifragales at a Larger-Sampling Scale. Genes 2023, 14, 694. https://doi.org/10.3390/genes14030694
Bi D, Han S, Zhou J, Zhao M, Zhang S, Kan X. Codon Usage Analyses Reveal the Evolutionary Patterns among Plastid Genes of Saxifragales at a Larger-Sampling Scale. Genes. 2023; 14(3):694. https://doi.org/10.3390/genes14030694
Chicago/Turabian StyleBi, De, Shiyun Han, Jun Zhou, Maojin Zhao, Sijia Zhang, and Xianzhao Kan. 2023. "Codon Usage Analyses Reveal the Evolutionary Patterns among Plastid Genes of Saxifragales at a Larger-Sampling Scale" Genes 14, no. 3: 694. https://doi.org/10.3390/genes14030694