

Genome-Wide Detection and Analysis of Copy Number Variation in Anhui Indigenous and Western Commercial Pig Breeds Using Porcine 80K SNP BeadChip
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Samples and Genotyping
2.3. Genome-Wide Detection of CNVs and CNVRs
2.4. Gene Contents and Functional Annotation
3. Results
3.1. Genome-Wide Detection of CNVs
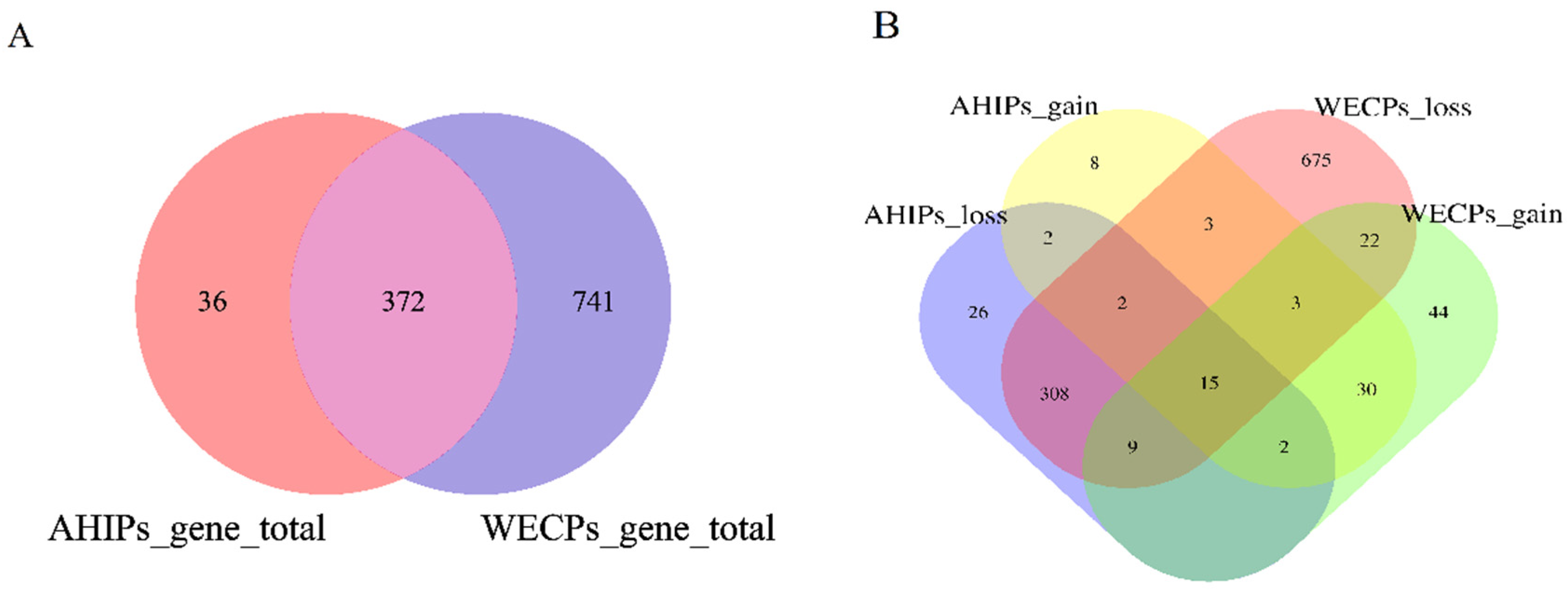
3.2. Analysis of Genes within AHIPs and WECPs CNVR
3.3. Enrichment Analysis of Candidate Genes in WECPs and AHIPs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iafrate, A.J.; Feuk, T.; Puymbroeck, L.V.; Rivera, M.N.; Lee, C. Detection of large-scale variation in the human genome. Nat. Genet. 2004, 36, 949–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Corbi-Botto, C.M.; Morales-Durand, H.; Zappa, M.E.; Sadaba, S.A.; Peral-García, P.; Giovambattista, G.; Díaz, S. Genomic structural diversity in Criollo Argentino horses: Analysis of copy number variations. Gene 2019, 695, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.; Madsen, O.; Megens, H.J.; Frantz, L.A.F.; Bosse, M.; Crooijmans, R.P.M.A.; Groenen, M.A.M. Copy number variation in the speciation of pigs: A possible prominent role for olfactory receptors. BMC Genom. 2015, 16, 330. [Google Scholar] [CrossRef] [Green Version]
- Lupski, J.R. Genomic rearrangements and sporadic disease. Nat. Genet. 2007, 39, S43–S47. [Google Scholar] [CrossRef]
- Lee, J.A.; Carvalho, C.M.; Lupski, J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 2007, 131, 1235–1247. [Google Scholar] [CrossRef] [Green Version]
- Redon, R.; Ishikawa, S.; Fitch, K.R.; Feuk, L.; Perry, G.H.; Andrews, T.D.; Fiegler, H.; Shapero, M.H.; Carson, A.R.; Chen, W.; et al. Global variation in copy number in the human genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef] [Green Version]
- Locke, M.E.; Milojevic, M.; Eitutis, S.T.; Patel, N.; Wishart, A.E.; Daley, M.; Hill, K.A. Genomic copy number variation in Mus musculus. BMC Genom. 2015, 16, 497. [Google Scholar] [CrossRef] [Green Version]
- Paudel, Y.; Madsen, O.; Megens, H.J.; Frantz, L.A.; Bosse, M.; Bastiaansen, J.W.; Crooijmans, R.P.; Groenen, M.A. Evolutionary dynamics of copy number variation in pig genomes in the context of adaptation and domestication. BMC Genom. 2013, 14, 449. [Google Scholar] [CrossRef] [Green Version]
- Bailey, J.A.; Kidd, J.M.; Eichler, E.E. Human copy number polymorphic genes. Cytogenet. Genome Res. 2008, 123, 234–243. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.J.; Dermitzakis, E.T.; Cox, T.; Smith, J.; Davies, R.; Banerjee, R.; Bonfield, J.; Mullikin, J.C.; Chung, Y.J.; Rogers, J.; et al. Complex haplotypes, copy number polymorphisms and coding variation in two recently divergent mouse strains. Nat. Genet. 2005, 37, 532–536. [Google Scholar] [CrossRef]
- Keel, B.N.; Lindholm-Perry, A.K.; Snelling, W.M. Evolutionary and Functional Features of Copy Number Variation in the Cattle Genome. Front. Genet. 2016, 7, 207. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, Z.; Sun, Y.; Wang, H.; Wang, C.; Yu, S.; Liu, J.; Zhang, Y.; Fan, B.; Li, K.; et al. Analysis of genome-wide copy number variations in Chinese indigenous and western pig breeds by 60 K SNP genotyping arrays. PLoS ONE 2014, 9, e106780. [Google Scholar] [CrossRef]
- Elferink, M.G.; Vallée, A.A.; Jungerius, A.P.; Crooijmans, R.P.; Groenen, M.A. Partial duplication of the PRLR and SPEF2 genes at the late feathering locus in chicken. BMC Genom. 2008, 9, 391. [Google Scholar] [CrossRef] [Green Version]
- Seo, B.Y.; Park, E.W.; Ahn, S.J.; Lee, S.H.; Jeon, J.T. An accurate method for quantifying and analyzing copy number variation in porcine KIT by an oligonucleotide ligation assay. BMC Genet. 2007, 8, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Jiang, J.; Yang, S.; Hou, Y.; Liu, G.E.; Zhang, S.; Zhang, Q.; Sun, D. CNV discovery for milk composition traits in dairy cattle using whole genome resequencing. BMC Genom. 2017, 18, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Zhao, P.; Yang, K.; Ning, C.; Wang, H.; Zhou, L.; Liu, J. CNV analysis of Meishan pig by next-generation sequencing and effects of AHR gene CNV on pig reproductive traits. J. Anim. Sci. Biotechnol. 2020, 11, 42. [Google Scholar] [CrossRef] [Green Version]
- Ran, X.Q.; Pan, H.; Huang, S.H.; Liu, C.; Niu, X.; Li, S.; Wang, J.F. Copy number variations of MTHFSD gene across pig breeds and its association with litter size traits in Chinese indigenous Xiang pig. J. Anim. Physiol. Anim. Nutr. 2018, 102, 1320–1327. [Google Scholar] [CrossRef]
- Revilla, M.; Puig-Oliveras, A.; Castello, A.; Crespo-Piazuelo, D.; Paludo, E.; Fernandez, A.I.; Ballester, M.; Folch, J.M. A global analysis of CNVs in swine using whole genome sequence data and association analysis with fatty acid composition and growth traits. PLoS ONE 2017, 12, e177014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Li, X.; Liu, J.; Zhang, W.; Zhou, M.; Wang, J.; Liu, L.; Su, S.; Zhao, F.; Chen, H.; et al. Genome-wide detection of genetic structure and runs of homozygosity analysis in Anhui indigenous and Western commercial pig breeds using PorcineSNP80k data. BMC Genom. 2022, 23, 373. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hadley, D.; Liu, R.; Glessner, J.; Grant, S.F.; Hakonarson, H.; Bucan, M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007, 17, 1665–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Jiang, J.; Fu, W.; Jiang, L.; Ding, X.; Liu, J.F.; Zhang, Q. A genome-wide detection of copy number variations using SNP genotyping arrays in swine. BMC Genom. 2012, 13, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Liu, G.E.; Bickhart, D.M.; Cardone, M.F.; Tassell, C.P.V. Genomic characteristics of cattle copy number variations. BMC Genom. 2011, 12, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Hu, H.J.; Yim, S.H.; Bae, J.S.; Kim, S.Y.; Chung, Y.J. CNVRuler: A copy number variation-based case-control association analysis tool. Bioinformatics 2012, 28, 1790–1792. [Google Scholar] [CrossRef] [Green Version]
- Kumar, H.; Panigrahi, M.; Saravanan, K.A.; Rajawat, D.; Parida, S.; Bhushan, B.; Gaur, G.K.; Dutt, T.; Mishra, B.P.; Singh, R.K. Genome-wide detection of copy number variations in Tharparkar cattle. Anim. Biotechnol. 2021, 1–8. [Google Scholar] [CrossRef]
- Kasprzyk, A. BioMart: Driving a paradigm change in biological data management. Database 2011, 2011, r49. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; Mccarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [Green Version]
- Bai, H.; He, Y.; Ding, Y.; Chu, Q.; Song, J. Genome-wide characterization of copy number variations in the host genome in genetic resistance to Marek’s disease using next generation sequencing. BMC Genet. 2020, 21, 1–12. [Google Scholar] [CrossRef]
- Wang, L.; Xu, L.; Liu, X.; Zhang, T.; Li, N.; Hay, E.H.; Zhang, Y.; Yan, H.; Zhao, K.; Liu, G.E. Copy number variation-based genome wide association study reveals additional variants contributing to meat quality in Swine. Sci. Rep. 2015, 5, 12535. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Chen, Q.; Liao, R.; Zhang, Z.; Zhang, X.; Liu, X.; Zhu, M.; Zhang, W.; Xue, M.; Yang, H.; et al. Genome-wide genetic variation discovery in Chinese Taihu pig breeds using next generation sequencing. Anim. Genet. 2017, 48, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Abernathy, J.; Li, X.; Jia, X.; Chou, W.; Lamont, S.J.; Crooijmans, R.; Zhou, H. Copy number variation in Fayoumi and Leghorn chickens analyzed using array comparative genomic hybridization. Anim. Genet. 2014, 45, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, T.; Wang, C. Detection and analysis of genome-wide copy number variation in the pig genome using an 80K SNP Beadchip. J. Anim. Breed. Genet. 2020, 137, 166–176. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. Human FOX gene family (Review). Int. J. Oncol. 2004, 25, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, H.; Peng, S.L. Forkhead transcription factors in immunology. Cell. Mol. Life Sci. 2005, 62, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Li, S.; Yang, F.; Cao, W.; Liu, H.; Feng, T.; Zhang, K.; Zhu, Z.; Liu, X.; Hu, Y.; et al. FoxJ1 inhibits African swine fever virus replication and viral S273R protein decreases the expression of FoxJ1 to impair its antiviral effect. Virol. Sin. 2022, 37, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lai, C.; Gaynor, S.R.B. Cloning of a cellular factor, interleukin binding factor, that binds to NFAT-like motifs in the human immunodeficiency virus long terminal repeat. Proc. Natl. Acad. Sci. USA 1991, 88, 7739–7743. [Google Scholar] [CrossRef] [Green Version]
- Nestal, D.M.G.; Carneiro, L.; Maia, R.C.; Lam, E.W.; Sharrocks, A.D. FOXK2 Transcription Factor and Its Emerging Roles in Cancer. Cancers 2019, 11, 393. [Google Scholar]
- Nirula, A.; Moore, D.J.; Gaynor, R.B. Constitutive Binding of the Transcription Factor Interleukin-2 (IL-2) Enhancer Binding Factor to the IL-2 Promoter. J. Biol. Chem. 1997, 272, 7736–7745. [Google Scholar] [CrossRef] [Green Version]
- Hackmann, K.; Stadler, A.; Schallner, J.; Franke, K.; Gerlach, E.M.; Schrock, E.; Rump, A.; Fauth, C.; Tinschert, S.; Oexle, K. Severe intellectual disability, West syndrome, Dandy-Walker malformation, and syndactyly in a patient with partial tetrasomy 17q25.3. Am. J. Med. Genet. A 2013, 161A, 3144–3149. [Google Scholar] [CrossRef]
- Fabian, J.; Dworschak, G.C.; Waffenschmidt, L.; Schierbaum, L.; Bendixen, C.; Heilmann-Heimbach, S.; Sivalingam, S.; Buness, A.; Schwarzer, N.; Boemers, T.M.; et al. Genome-wide identification of disease-causing copy number variations in 450 individuals with anorectal malformations. Eur. J. Hum. Genet. 2022, 31, 105–111. [Google Scholar] [CrossRef]
- Boschmann, S.E.; Goeldner, I.; Tuon, F.F.; Schiel, W.; Aoyama, F.; de Messias-Reason, I.J. Mannose-binding lectin polymorphisms and rheumatoid arthritis: A short review and meta-analysis. Mol. Immunol. 2016, 69, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Bergman, I.M.; Edman, K.; van As, P.; Huisman, A.; Juul-Madsen, H.R. A two-nucleotide deletion renders the mannose-binding lectin 2 (MBL2) gene nonfunctional in Danish Landrace and Duroc pigs. Immunogenetics 2014, 66, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Gao, P. Sensitization to Cockroach Allergen: Immune Regulation and Genetic Determinants. Clin. Dev. Immunol. 2012, 2012, 563760. [Google Scholar] [CrossRef] [Green Version]
- Stravalaci, M.; Pagani, I.; Paraboschi, E.M.; Pedotti, M.; Doni, A.; Scavello, F.; Mapelli, S.N.; Sironi, M.; Perucchini, C.; Varani, L. Recognition and inhibition of SARS-CoV-2 by humoral innate immunity pattern recognition molecules. Nat. Immunol. 2022, 23, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, I.; Fernandez, I.; Soudre, A.; Traore, A.; Perez-Pardal, L.; Sanou, M.; Tapsoba, S.; Menendez-Arias, N.A.; Goyache, F. Identification of genomic regions and candidate genes of functional importance for gastrointestinal parasite resistance traits in Djallonke sheep of Burkina Faso. Arch. Anim. Breed. 2019, 62, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Webb, G.J.; Hirschfield, G.M.; Lane, P.J. OX40, OX40L and Autoimmunity: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2016, 50, 312–332. [Google Scholar] [CrossRef] [PubMed]
- Aspeslagh, S.; Postel-Vinay, S.; Rusakiewicz, S.; Soria, J.C.; Zitvogel, L.; Marabelle, A. Rationale for anti-OX40 cancer immunotherapy. Eur. J. Cancer 2016, 52, 50–66. [Google Scholar] [CrossRef]
- Bell, R.B.; Leidner, R.S.; Crittenden, M.R.; Curti, B.D.; Feng, Z.; Montler, R.; Gough, M.J.; Fox, B.A.; Weinberg, A.D.; Urba, W.J. OX40 signaling in head and neck squamous cell carcinoma: Overcoming immunosuppression in the tumor microenvironment. Oral Oncol. 2016, 52, 1–10. [Google Scholar] [CrossRef]
- Wang, D.; Hu, H.; Ding, H.; Chi, Q.; Tian, F.; Zhao, H. Elevated expression of TNFRSF4 impacts immune cell infiltration and gene mutation in hepatocellular carcinoma. Cancer Biomark. 2022, 36, 147–159. [Google Scholar] [CrossRef]
- Jiao, F.; Gong, Z. The Beneficial Roles of SIRT1 in Neuroinflammation-Related Diseases. Oxidative Med. Cell. Longev. 2020, 2020, 6782872. [Google Scholar] [CrossRef]
- Lei, Y.; Wang, J.; Wang, D.; Li, C.; Liu, B.; Fang, X.; You, J.; Guo, M.; Lu, X.Y. SIRT1 in forebrain excitatory neurons produces sexually dimorphic effects on depression-related behaviors and modulates neuronal excitability and synaptic transmission in the medial prefrontal cortex. Mol. Psychiatr. 2020, 25, 1094–1111. [Google Scholar] [CrossRef] [Green Version]
- Daenthanasanmak, A.; Iamsawat, S.; Chakraborty, P.; Nguyen, H.D.; Bastian, D.; Liu, C.; Mehrotra, S.; Yu, X.Z. Targeting Sirt-1 controls GVHD by inhibiting T-cell allo-response and promoting Treg stability in mice. Blood 2019, 133, 266–279. [Google Scholar] [CrossRef] [Green Version]
- Labiner, H.E.; Sas, K.M.; Baur, J.A.; Sims, C.A. Sirtuin 1 deletion increases inflammation and mortality in sepsis. J. Trauma Acute Care Surg. 2022, 93, 672–678. [Google Scholar] [CrossRef]
- Li, M.; Zhang, W.; Zhang, J.; Li, X.; Zhang, F.; Zhu, W.; Meng, L.; Holmdahl, R.; Lu, S. Ncf1 Governs Immune Niches in the Lung to Mediate Pulmonary Inflammation in Mice. Front. Immunol. 2021, 12, 783944. [Google Scholar] [CrossRef]
- Li, X.J.; Liu, L.Q.; Dong, H.; Yang, J.J.; Wang, W.W.; Zhang, Q.; Wang, C.L.; Zhou, J.; Chen, H.Q. Comparative genome-wide methylation analysis of longissimus dorsi muscles in Yorkshire and Wannanhua pigs. Anim. Genet. 2021, 52, 78–89. [Google Scholar] [CrossRef]
- Hu, H.; Wu, C.; Ding, Y.; Zhang, X.; Yin, Z. Comparative analysis of meat sensory quality, antioxidant status, growth hormone and orexin between Anqingliubai and Yorkshire pigs. J. Appl. Anim. Res. 2019, 47, 357–361. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wang, C.; Jin, E.; Gu, Y.; Li, S.; Li, Q. Identification of differentially expressed genes in longissimus dorsi muscle between Wei and Yorkshire pigs using RNA sequencing. Genes Genom. 2018, 40, 413–421. [Google Scholar] [CrossRef]
- Cui, J.X.; Zeng, Q.F.; Chen, W.; Zhang, H.; Zeng, Y.Q. Analysis and preliminary validation of the molecular mechanism of fat deposition in fatty and lean pigs by high-throughput sequencing. Mamm. Genome 2019, 30, 71–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.X.; Yang, Z.P.; Shi, X.K.; Li, J.Y.; Ji, D.J.; Mao, Y.J.; Gao, L.L.C.J. Association of SCD1 and DGAT1 SNPs with the intramuscular fat traits in Chinese Simmental cattle and their distribution in eight Chinese cattle breeds. Mol. Biol. Rep. 2012, 39, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Dai, R.; Zhou, H.; Fang, Q.; Zhou, P.; Yang, Y.; Jiang, S.; Hickford, J. Variation in Ovine DGAT1 and Its Association with Carcass Muscle Traits in Southdown Sheep. Genes 2022, 13, 1670. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Xu, D.; Zuo, B.; Lei, M.; Xiong, Y.; Chen, H.; Zhou, Y.; Wu, X. Ectopic overexpression of porcine DGAT1 increases intramuscular fat content in mouse skeletal muscle. Transgenic Res. 2013, 22, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Cheng, J.; Chen, Y.; Yang, L.; Raza, S.; Huang, Y.; Lei, C.; Liu, G.E.; Lan, X.; Chen, H. Distribution of DGAT1 copy number variation in Chinese goats and its associations with milk production traits. Anim. Biotechnol. 2021, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Guo, Y.; Liu, S.; Meng, Q. Genome-Wide Assessment Characteristics of Genes Overlapping Copy Number Variation Regions in Duroc Purebred Population. Front. Genet. 2021, 12, 1964. [Google Scholar] [CrossRef]
- Maga, J.A. Flavor potentiators. Crit. Rev. Food Sci. Nutr. 1983, 18, 231–312. [Google Scholar] [CrossRef]
- Bogan, K.L.; Brenner, C. 5′-Nucleotidases and their new roles in NAD+ and phosphate metabolism. New J. Chem. 2010, 34, 845. [Google Scholar] [CrossRef]
- Surette, M.E.; Gill, T.A.; Leblanc, P.J. Biochemical basis of postmortem nucleotide catabolism in cod (Gadus morhua) and its relationship to spoilage. J. Agric. Food Chem. 1988, 36, 19–22. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Yoshikawa, T.; Ikeda, S.; Ninomiya, T. Measurement of the Relative Taste Intensity of Some L--Amino Acids And 5′-Nucleotides. J. Food Sci. 1971, 36, 846–849. [Google Scholar] [CrossRef]
- Uemoto, Y.; Ohtake, T.; Sasago, N.; Takeda, M.; Abe, T.; Sakuma, H.; Kojima, T.; Sasaki, S. Effect of two non-synonymous ecto-5′-nucleotidase variants on the genetic architecture of inosine 5′-monophosphate (IMP) and its degradation products in Japanese Black beef. BMC Genom. 2017, 18, 874. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, T.; Toita, T.; Uemoto, Y. Estimates of genetic parameters for adenosine triphosphate-related compounds at different aging periods and NT5E genotypes in Japanese Black beef. Anim. Sci. J. 2022, 93, e13748. [Google Scholar] [CrossRef]
- Ge, K.; Chen, X.; Kuang, J.; Yang, L.; Geng, Z. Comparison of liver transcriptome from high- and low-intramuscular fat Chaohu ducks provided additional candidate genes for lipid selection. 3 Biotech 2019, 9, 251. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Sample Size | Total Number of CNV | Unique Number of CNVR | Gain | Loss | Average Size (kb) | Max Size (kb) | Median Size (kb) | Min Size (kb) |
|---|---|---|---|---|---|---|---|---|---|
| AHIPs | 170 | 3863 | 225 | 47 | 178 | 1187.45 | 6400.56 | 774.74 | 25.44 |
| WECPs | 150 | 7546 | 379 | 86 | 293 | 1459.32 | 9761.85 | 901.97 | 4.85 |
| Total | 320 | 11409 | 604 | 133 | 471 | 1358.04 | 9761.85 | 4.85 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, C.; Zhang, W.; Jiang, Y.; Zhou, M.; Liu, L.; Su, S.; Li, X.; Wang, C. Genome-Wide Detection and Analysis of Copy Number Variation in Anhui Indigenous and Western Commercial Pig Breeds Using Porcine 80K SNP BeadChip. Genes 2023, 14, 654. https://doi.org/10.3390/genes14030654
Xu C, Zhang W, Jiang Y, Zhou M, Liu L, Su S, Li X, Wang C. Genome-Wide Detection and Analysis of Copy Number Variation in Anhui Indigenous and Western Commercial Pig Breeds Using Porcine 80K SNP BeadChip. Genes. 2023; 14(3):654. https://doi.org/10.3390/genes14030654
Chicago/Turabian StyleXu, Chengliang, Wei Zhang, Yao Jiang, Mei Zhou, Linqing Liu, Shiguang Su, Xueting Li, and Chonglong Wang. 2023. "Genome-Wide Detection and Analysis of Copy Number Variation in Anhui Indigenous and Western Commercial Pig Breeds Using Porcine 80K SNP BeadChip" Genes 14, no. 3: 654. https://doi.org/10.3390/genes14030654