
The Developmental Origins of Cancer: A Review of the Genes Expressed in Embryonic Cells with Implications for Tumorigenesis

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pluripotency Gene Regulatory Network
3. The p53 Pathway
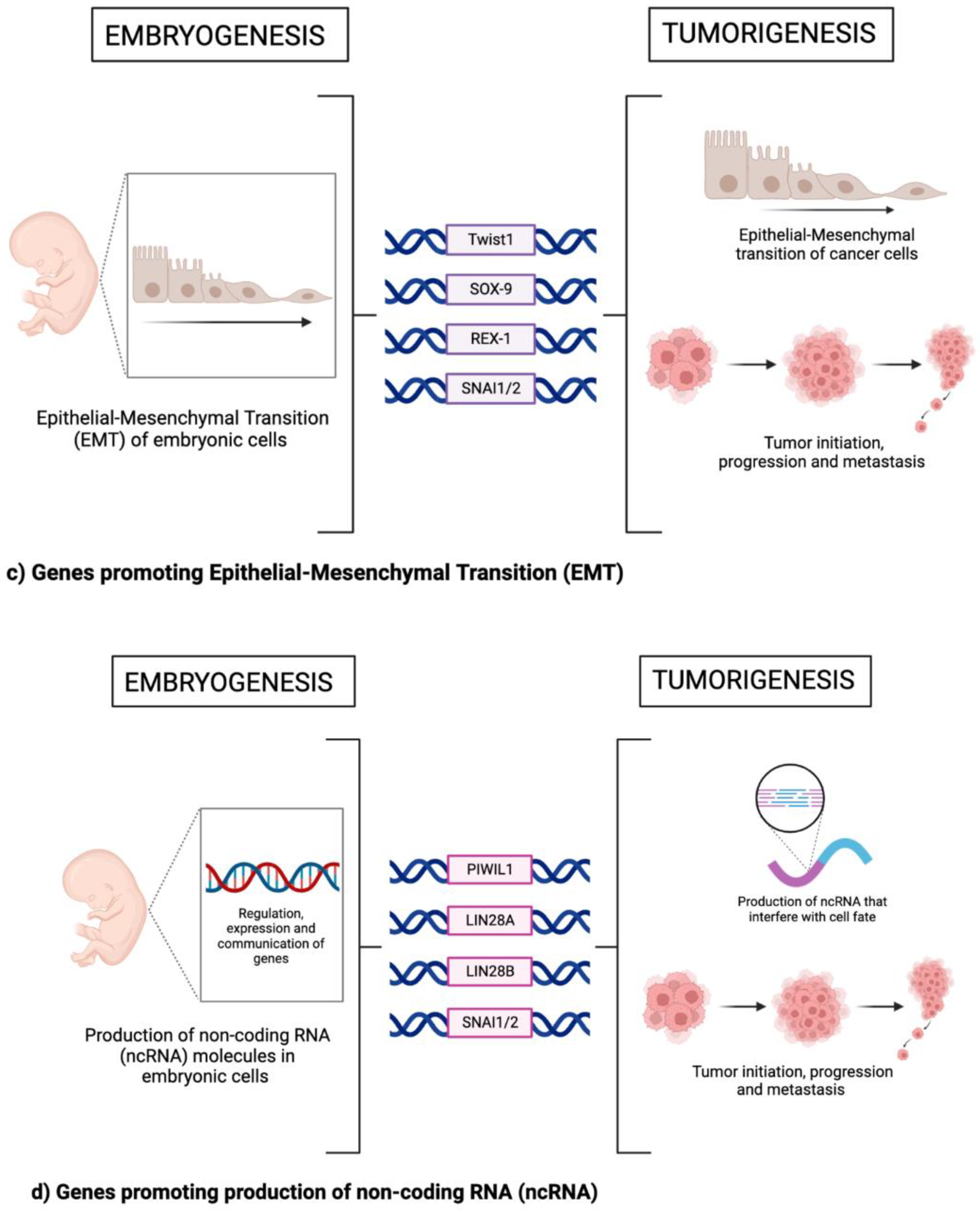
4. Epithelial Mesenchymal Transition (EMT)
5. Non-Coding RNAs
6. Clinical Relevance
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Biswas, A.; Hutchins, R. Embryonic Stem Cells. Stem Cells Dev. 2007, 16, 213–222. [Google Scholar] [CrossRef]
- Kim, J.; Orkin, S.H. Embryonic stem cell-specific signatures in cancer: Insights into genomic regulatory networks and implications for medicine. Genome Med. 2011, 3, 75–78. [Google Scholar] [CrossRef] [Green Version]
- Manzo, G. Similarities Between Embryo Development and Cancer Process Suggest New Strategies for Research and Therapy of Tumors: A New Point of View. Front. Cell Dev. Biol. 2019, 7, 20. [Google Scholar] [CrossRef]
- Kar, S.; Patra, S.K. Overexpression of OCT4 induced by modulation of histone marks plays crucial role in breast cancer progression. Gene 2018, 643, 35–45. [Google Scholar] [CrossRef]
- Lu, C.-S.; Shieh, G.-S.; Wang, C.-T.; Su, B.-H.; Su, Y.-C.; Chen, Y.-C.; Su, W.-C.; Wu, P.; Yang, W.-H.; Shiau, A.-L.; et al. Chemotherapeutics-induced Oct4 expression contributes to drug resistance and tumor recurrence in bladder cancer. Oncotarget 2016, 8, 30844–30858. [Google Scholar] [CrossRef] [Green Version]
- Villodre, E.S.; Kipper, F.C.; Pereira, M.B.; Lenz, G. Roles of OCT4 in tumorigenesis, cancer therapy resistance and prognosis. Cancer Treat. Rev. 2016, 51, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rizzino, A.; Wuebben, E.L. Sox2/Oct4: A delicately balanced partnership in pluripotent stem cells and embryogenesis. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2016, 1859, 780–791. [Google Scholar] [CrossRef] [PubMed]
- de Wet, L.; Williams, A.; Gillard, M.; Kregel, S.; Lamperis, S.; Gutgesell, L.C.; Vellky, J.E.; Brown, R.; Conger, K.; Paner, G.P.; et al. SOX2 mediates metabolic reprogramming of prostate cancer cells. Oncogene 2022, 41, 1190–1202. [Google Scholar] [CrossRef]
- Xie, X.; Piao, L.; Cavey, G.S.; Old, M.; Teknos, T.N.; Mapp, A.K.; Pan, Q. Phosphorylation of Nanog is essential to regulate Bmi1 and promote tumorigenesis. Oncogene 2013, 33, 2040–2052. [Google Scholar] [CrossRef] [Green Version]
- Saga, K.; Park, J.; Nimura, K.; Kawamura, N.; Ishibashi, A.; Nonomura, N.; Kaneda, Y. NANOG helps cancer cells escape NK cell attack by downregulating ICAM1 during tumorigenesis. J. Exp. Clin. Cancer Res. 2019, 38, 416. [Google Scholar] [CrossRef] [PubMed]
- Luk, S.T.; Ng, K.; Zhou, L.; Tong, M.; Wong, T.; Yu, H.; Lo, C.; Man, K.; Guan, X.; Lee, T.K.; et al. Deficiency in embryonic stem cell marker reduced expression 1 activates mitogen-activated protein kinase kinase 6–dependent p38 mitogen-activated protein kinase signaling to drive hepatocarcinogenesis. Hepatology 2020, 72, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, D.; Xu, J.; Wang, Y.; Ma, F.; Li, Z.; Liu, N. Stat3 activation is critical for pluripotency maintenance. J. Cell. Physiol. 2018, 234, 1044–1051. [Google Scholar] [CrossRef]
- Yang, J.; van Oosten, A.L.; Theunissen, T.W.; Guo, G.; Silva, J.C.; Smith, A. Stat3 Activation Is Limiting for Reprogramming to Ground State Pluripotency. Cell Stem Cell 2010, 7, 319–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Oosten, A.L.; Costa, Y.; Smith, A.; Silva, J.C. JAK/STAT3 signalling is sufficient and dominant over antagonistic cues for the establishment of naive pluripotency. Nat. Commun. 2012, 3, 817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raz, R.; Lee, C.-K.; Cannizzaro, L.A.; D’Eustachio, P.; Levy, D.E. Essential role of STAT3 for embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. USA 1999, 96, 2846–2851. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, R.K.; Beattie, G.M.; Lopez, A.D.; Bucay, N.; King, C.C.; Firpo, M.T.; Rose-John, S.; Hayek, A. Maintenance of Pluripotency in Human Embryonic Stem Cells Is STAT3 Independent. Stem Cells 2004, 22, 522–530. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Torigoe, S.E.; Xiao, J.; Mai, D.H.; Li, L.; Davis, F.P.; Dong, P.; Marie-Nelly, H.; Grimm, J.; Lavis, L.; et al. A dynamic interplay of enhancer elements regulates Klf4 expression in naïve pluripotency. Genes Dev. 2017, 31, 1795–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmerman, D.; Remmers, T.; Hillenius, S.; Looijenga, L. Mechanisms of TP53 pathway inactivation in embryonic and somatic cells—Relevance for understanding (germ cell) tumorigenesis. Int. J. Mol. Sci. 2021, 22, 5377. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Murphy, M.E. Genetic modifiers of the p53 pathway. Cold Spring Harb. Perspect. Med. 2016, 6, a026302. [Google Scholar] [CrossRef]
- Mogi, A.; Kuwano, H. TP53 mutations in nonsmall cell lung cancer. J. Biomed. Biotechnol. 2011, 2011, 583929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Liu, N.; Liu, J.; Liu, Y.; Zhang, C.; Long, S.; Luo, G.; Zhang, L.; Zhang, Y. Mutant p53 drives cancer chemotherapy resistance due to loss of function on activating transcription of PUMA. Cell Cycle 2019, 18, 3442–3455. [Google Scholar] [CrossRef] [PubMed]
- Subhasree, N.; Jiangjiang, Q.; Kalkunte, S.; Minghai, W.; Ruiwen, Z. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Ramos, F.S.; Wons, L.; Cavalli, I.J.; Ribeiro, E.M. Epithelial-mesenchymal transition in cancer: An overview. Integr. Cancer Sci. Ther. 2017, 4, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Rahman, M.A.; Chen, Z.G.; Shin, D.M. Multiple biological functions of Twist1 in various cancers. Oncotarget 2017, 8, 20380–20393. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Li, G.; Huang, S.; Feng, Y.; Zhou, A. SOX9 promotes epithelial-mesenchymal transition via the Hippo-YAP signaling pathway in gastric carcinoma cells. Oncol. Lett. 2019, 18, 599–608. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhang, Z.; Yu, X.; Huang, X.; Liu, Z.; Chai, Y.; Yang, L.; Wang, Q.; Li, M.; Zhao, J.; et al. Unbalanced YAP–SOX9 circuit drives stemness and malignant progression in esophageal squamous cell carcinoma. Oncogene 2018, 38, 2042–2055. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.-T.; Liu, X.-F.; Yang, W.-T.; Zheng, P.-S. REX1 promotes EMT-induced cell metastasis by activating the JAK2/STAT3-signaling pathway by targeting SOCS1 in cervical cancer. Oncogene 2019, 38, 6940–6957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maturi, V.; Morén, A.; Enroth, S.; Heldin, C.; Moustakas, A. Genomewide binding of transcription factor Snail1 in triple-negative breast cancer cells. Mol. Oncol. 2018, 12, 1153–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, W.; Miyazaki, K.; Kitajima, Y. Inverse correlation between E-cadherin and Snail expression in hepatocellular carcinoma cell lines in vitro and in vivo. Br. J. Cancer 2002, 86, 98–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elloul, S.; Elstrand, M.B.; Nesland, J.M.; Tropé, C.G.; Kvalheim, G.; Goldberg, I.; Reich, R.; Davidson, B. Snail, Slug, and Smad-interacting protein 1 as novel parameters of disease aggressiveness in metastatic ovarian and breast carcinoma. Cancer 2005, 103, 1631–1643. [Google Scholar] [CrossRef]
- Alves, C.L.; Elias, D.; Lyng, M.B.; Bak, M.; Ditzel, H.J. SNAI2 upregulation is associated with an aggressive phenotype in fulvestrant-resistant breast cancer cells and is an indicator of poor response to endocrine therapy in estrogen receptor-positive metastatic breast cancer. Breast Cancer Res. 2018, 20, 60. [Google Scholar] [CrossRef] [Green Version]
- Masuo, K.; Chen, R.; Yogo, A.; Sugiyama, A.; Fukuda, A.; Masui, T.; Uemoto, S.; Seno, H.; Takaishi, S. SNAIL2 contributes to tumorigenicity and chemotherapy resistance in pancreatic cancer by regulating IGFBP2. Cancer Sci. 2021, 112, 4987–4999. [Google Scholar] [CrossRef]
- Hu, Y.; Dai, M.; Zheng, Y.; Wu, J.; Yu, B.; Zhang, H.; Kong, W.; Wu, H.; Yu, X. Epigenetic suppression of E-cadherin expression by Snail2 during the metastasis of colorectal cancer. Clin. Epigenetics 2018, 10, 154. [Google Scholar] [CrossRef]
- Taki, M.; Abiko, K.; Baba, T.; Hamanishi, J.; Yamaguchi, K.; Murakami, R.; Yamanoi, K.; Horikawa, N.; Hosoe, Y.; Nakamura, E.; et al. Snail promotes ovarian cancer progression by recruiting myeloid-derived suppressor cells via CXCR2 ligand upregulation. Nat. Commun. 2018, 9, 1685. [Google Scholar] [CrossRef] [Green Version]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef]
- Pauli, A.; Rinn, J.; Schier, A.F. Non-coding RNAs as regulators of embryogenesis. Nat. Rev. Genet. 2011, 12, 136–149. [Google Scholar] [CrossRef]
- Han, Y.-N.; Li, Y.; Xia, S.-Q.; Zhang, Y.-Y.; Zheng, J.-H.; Li, W. Piwi proteins and piwi-interacting RNA: Emerging roles in cancer. Cell. Physiol. Biochem. 2017, 44, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, A.; Tejero, R.; Viñolas, N.; Cordeiro, A.; Marrades, R.M.; Fuster, D.; Caritg, O.; Moises, J.; Muñoz, C.; Molins, L.; et al. The significance of PIWI family expression in human lung embryogenesis and non-small cell lung cancer. Oncotarget 2015, 6, 31544–31556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, K.; Zhang, K.; Kong, J.; Wang, C.; Gu, Y.; Liang, C.; Jiang, T.; Qin, N.; Liu, J.; Guo, X.; et al. Cancer-testis gene PIWIL1 promotes cell proliferation, migration, and invasion in lung adenocarcinoma. Cancer Med. 2017, 7, 157–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erber, R.; Meyer, J.; Taubert, H.; Fasching, P.A.; Wach, S.; Häberle, L.; Gaß, P.; Schulz-Wendtland, R.; Landgraf, L.; Olbricht, S.; et al. PIWI-Like 1 and PIWI-Like 2 Expression in Breast Cancer. Cancers 2020, 12, 2742. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Yang, Z.-Z.; Liu, S.; Yang, F.; Lin, H. PIWIL1 promotes gastric cancer via a piRNA-independent mechanism. Proc. Natl. Acad. Sci. USA 2020, 117, 22390–22401. [Google Scholar] [CrossRef]
- Chu, H.; Xia, L.; Qiu, X.; Gu, D.; Zhu, L.; Jin, J.; Hui, G.; Hua, Q.; Du, M.; Tong, N.; et al. Genetic variants in noncoding PIWI-interacting RNA and colorectal cancer risk. Cancer 2015, 121, 2044–2052. [Google Scholar] [CrossRef]
- Chen, C.; Liu, J.; Xu, G. Overexpression of PIWI proteins in human stage III epithelial ovarian cancer with lymph node metastasis. Cancer Biomark. 2013, 13, 315–321. [Google Scholar] [CrossRef]
- Eckstein, M.; Jung, R.; Weigelt, K.; Sikic, D.; Stöhr, R.; Geppert, C.; Agaimy, A.; Lieb, V.; Hartmann, A.; Wullich, B.; et al. Piwi-like 1 and -2 protein expression levels are prognostic factors for muscle invasive urothelial bladder cancer patients. Sci. Rep. 2018, 8, 17693. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, K.; Wu, Q.; Jin, C.-S.; Yuan, H.-J.; Cheng, J.-Z. Long non-coding RNA HNF1A-AS is upregulated and promotes cell proliferation and metastasis in nasopharyngeal carcinoma. Cancer Biomark. 2016, 16, 291–300. [Google Scholar] [CrossRef]
- Middelkamp, M.; Ruck, L.; Krisp, C.; Sumisławski, P.; Mohammadi, B.; Dottermusch, M.; Meister, V.; Küster, L.; Schlüter, H.; Windhorst, S.; et al. Overexpression of Lin28A in neural progenitor cells in vivo does not lead to brain tumor formation but results in reduced spine density. Acta Neuropathol. Commun. 2021, 9, 185. [Google Scholar] [CrossRef]
- Qiu, C.; Ma, Y.; Wang, J.; Peng, S.; Huang, Y. Lin28-mediated post-transcriptional regulation of Oct4 expression in human embryonic stem cells. Nucleic Acids Res. 2009, 38, 1240–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balzeau, J.; Menezes, M.R.; Cao, S.; Hagan, J.P. The LIN28/let-7 Pathway in Cancer. Front. Genet. 2017, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Pretzsch, E.; Max, N.; Kirchner, T.; Engel, J.; Werner, J.; Klauschen, F.; Angele, M.; Neumann, J. LIN28 promotes tumorigenesis in colorectal cancer but is not associated with metastatic spread. Pathol.—Res. Pract. 2021, 228, 153669. [Google Scholar] [CrossRef] [PubMed]
- Guilharducci, R.L.; Xavier, P.L.P.; da Silveira, J.C.; Cordeiro, Y.D.G.; Biondi, L.R.; Strefezzi, R.D.F.; Fukumasu, H. Expression of LIN28A/B and Let-7 miRNAs in canine mammary carcinomas. Cienc. Rural. 2022, 52, e20210171. [Google Scholar] [CrossRef]
- Skrzypek, K.; Majka, M. Interplay among snail transcription factor, MicroRNAs, long non-coding RNAS, and circular RNAS in the regulation of tumor growth and metastasis. Cancers 2020, 12, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Sun, Q.; Zhang, J.; Yu, J.; Chen, W.; Zhang, Z. Downregulation of miR-153 contributes to epithelial-mesenchymal transition and tumor metastasis in human epithelial cancer. Carcinog. 2012, 34, 539–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Fu, T.; Zhang, L. MicroRNA-153 suppresses human laryngeal squamous cell carcinoma migration and invasion by targeting the SNAI1 gene. Oncol. Lett. 2018, 16, 5075–5083. [Google Scholar] [CrossRef]
- Luan, W.; Shi, Y.; Zhou, Z.; Xia, Y.; Wang, J. circRNA_0084043 promote malignant melanoma progression via miR-153-3p/Snail axis. Biochem. Biophys. Res. Commun. 2018, 502, 22–29. [Google Scholar] [CrossRef]
- Zhao, G.; Zhang, Y.; Zhao, Z.; Cai, H.; Zhao, X.; Yang, T.; Chen, W.; Yao, C.; Wang, Z.; Wang, Z.; et al. MiR-153 reduces stem cell-like phenotype and tumor growth of lung adenocarcinoma by targeting Jagged1. Stem Cell Res. Ther. 2020, 11, 170. [Google Scholar] [CrossRef]
- Zuo, Z.; Ye, F.; Liu, Z.; Huang, J.; Gong, Y. MicroRNA-153 inhibits cell proliferation, migration, invasion and epithelial-mesenchymal transition in breast cancer via direct targeting of RUNX2. Exp. Ther. Med. 2019, 17, 4693–4702. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, B.; Li, W.; Fu, L.; Zhu, Z.; Dong, J.-T. Epigenetic silencing of mir-203 upregulates snai2 and contributes to the invasiveness of malignant breast cancer cells. Genes Cancer 2011, 2, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.H.; Robinton, D.A.; Seligson, M.T.; Wu, L.; Li, L.; Rakheja, D.; Comerford, S.A.; Ramezani, S.; Sun, X.; Parikh, M.S.; et al. LIN28B is sufficient to drive liver cancer and necessary for its maintenance in murine models. Cancer Cell 2014, 26, 248–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penson, A.; Camacho, N.; Zheng, Y.; Varghese, A.M.; Al-Ahmadie, H.; Razavi, P.; Chandarlapaty, S.; Vallejo, C.E.; Vakiani, E.; Gilewski, T.; et al. Development of genome-derived tumor type prediction to inform clinical cancer care. JAMA Oncol. 2020, 6, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Barh, D.; Malhotra, R.; Ravi, B.; Sindhurani, P. Microrna let-7: An emerging next-generation cancer therapeutic. Curr. Oncol. 2010, 17, 70–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavares, M.O.; Milan, T.M.; Bighetti-Trevisan, R.L.; Leopoldino, A.M.; de Almeida, L.O. Pharmacological inhibition of HDAC6 overcomes cisplatin chemoresistance by targeting cancer stem cells in oral squamous cell carcinoma. J. Oral Pathol. Med. 2022, 51, 529–537. [Google Scholar] [CrossRef]
- Fan, G.-T.; Ling, Z.-H.; He, Z.-W.; Wu, S.-J.; Zhou, G.-X. Suppressing CHD1L reduces the proliferation and chemoresistance in osteosarcoma. Biochem. Biophys. Res. Commun. 2021, 554, 214–221. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balachandran, S.; Narendran, A. The Developmental Origins of Cancer: A Review of the Genes Expressed in Embryonic Cells with Implications for Tumorigenesis. Genes 2023, 14, 604. https://doi.org/10.3390/genes14030604
Balachandran S, Narendran A. The Developmental Origins of Cancer: A Review of the Genes Expressed in Embryonic Cells with Implications for Tumorigenesis. Genes. 2023; 14(3):604. https://doi.org/10.3390/genes14030604
Chicago/Turabian StyleBalachandran, Savitha, and Aru Narendran. 2023. "The Developmental Origins of Cancer: A Review of the Genes Expressed in Embryonic Cells with Implications for Tumorigenesis" Genes 14, no. 3: 604. https://doi.org/10.3390/genes14030604