
Production and Characterization of K562 Cellular Clones Hyper-Expressing the Gene Encoding α-Globin: Preliminary Analysis of Biomarkers Associated with Autophagy
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture of Human Erythroleukemic K562 Cells (K562 Wild Type) and Obtained Clones
2.2. Production of K562 Clones Hyper-Expressing α-Globin
2.3. Cell Proliferation Analysis and Apoptosis Profile
2.4. RNA Extraction
2.5. RT-qPCR Analysis
2.6. Western Blotting Analysis
2.7. FACS Analysis
2.8. Genomic DNA Isolation from Whole Blood or Cultured Cells
2.9. Agarose Gel Electrophoresis
2.10. Statistical Analysis
3. Results
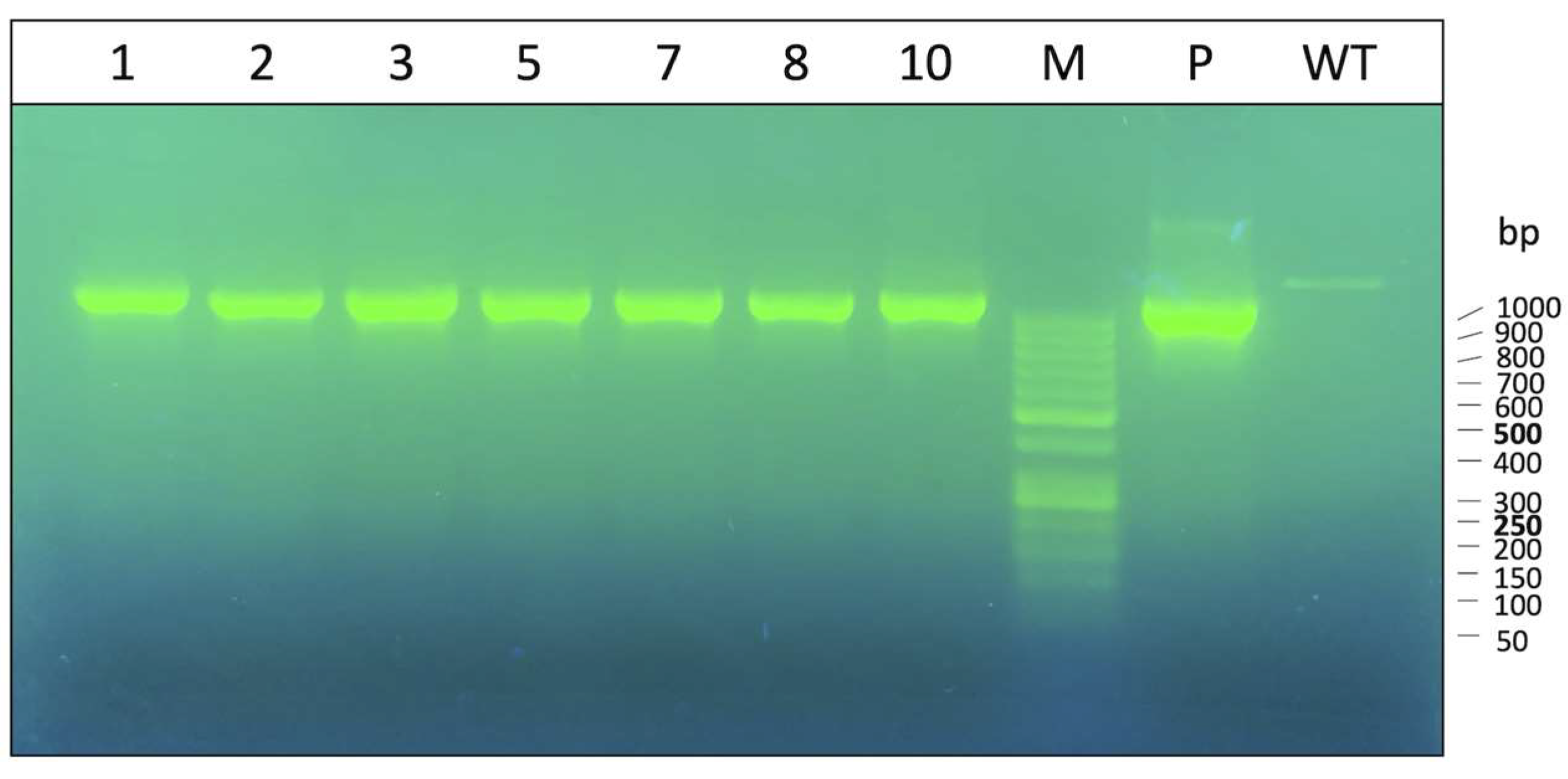
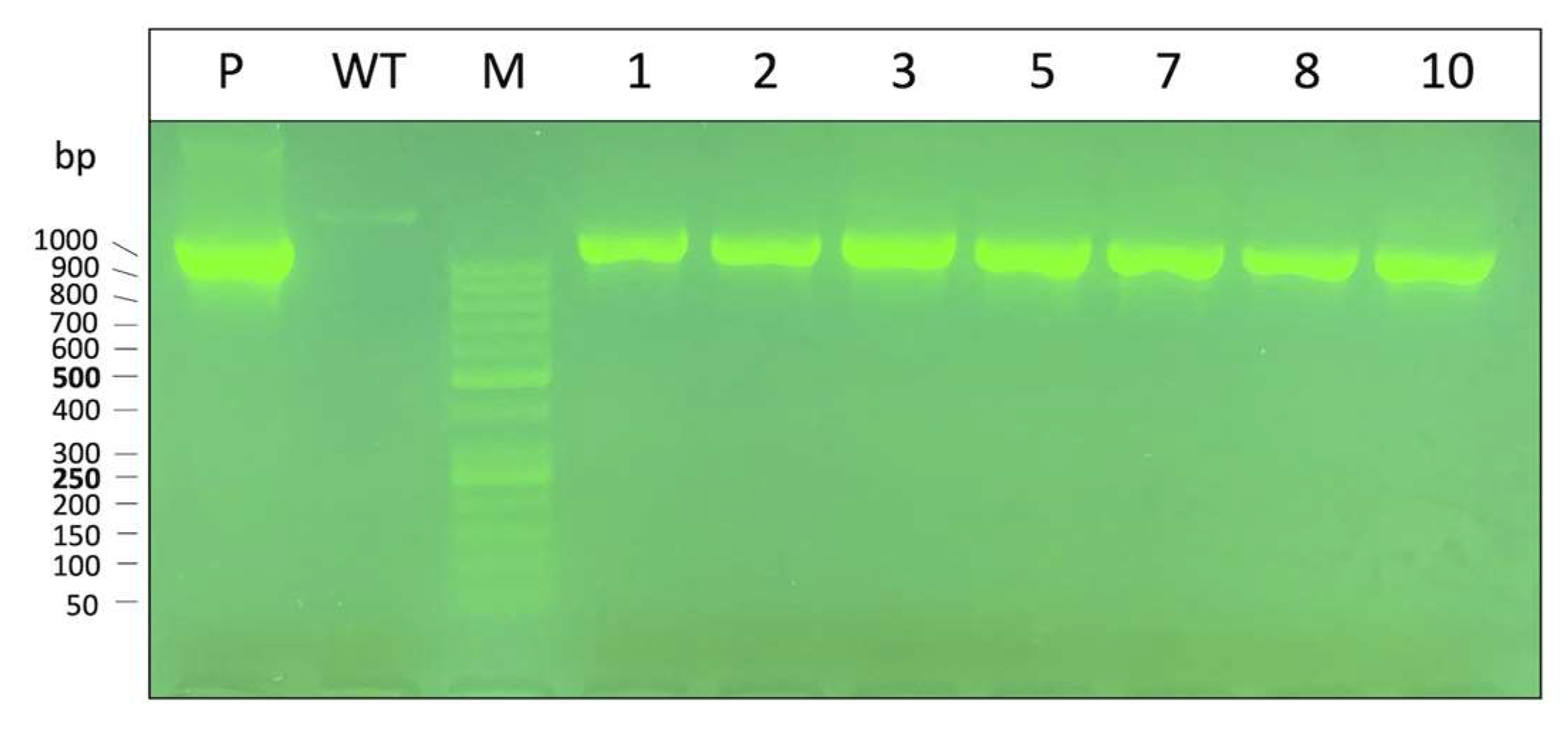
3.1. Characterization of Obtained K562 Clones
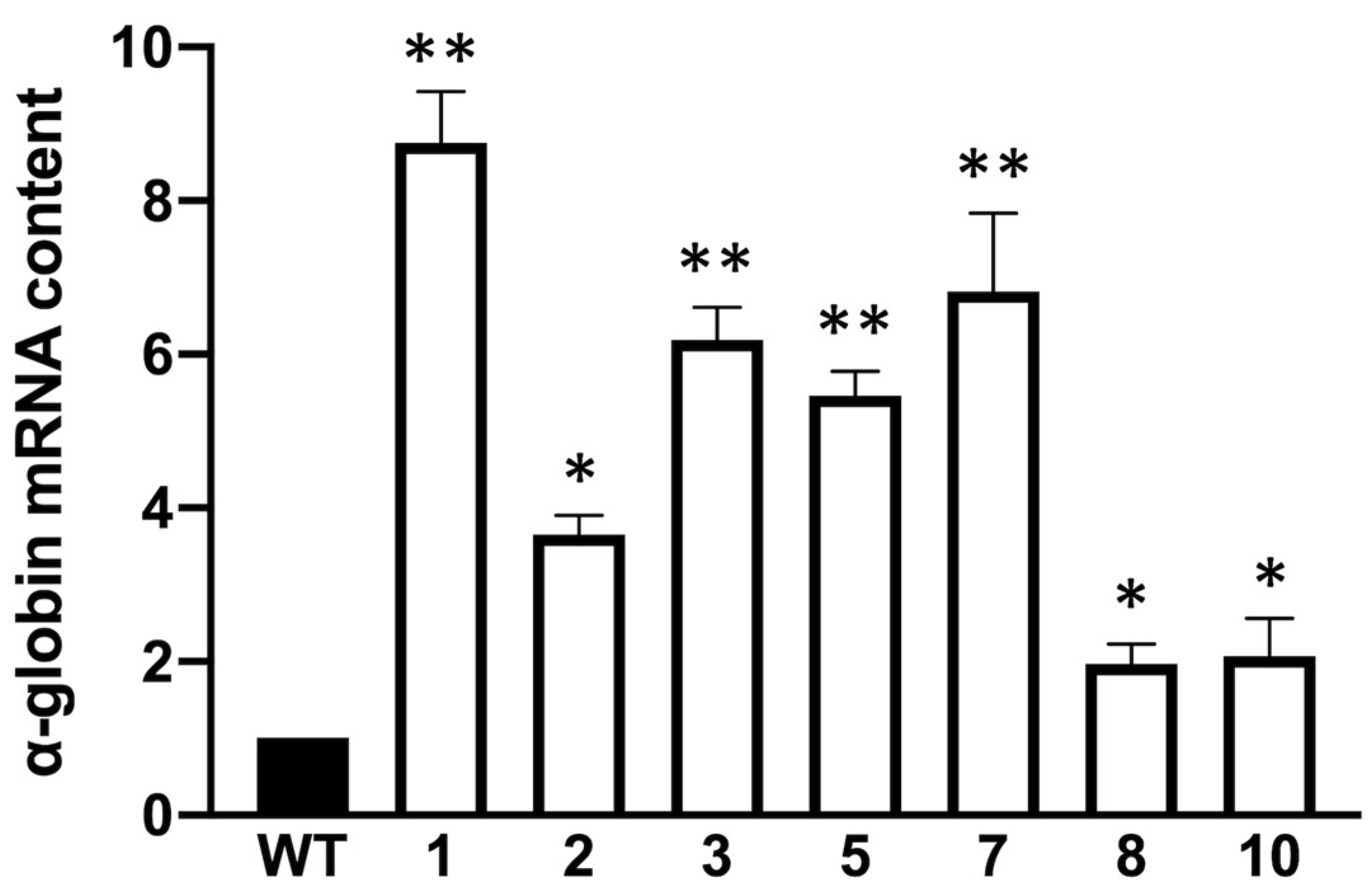
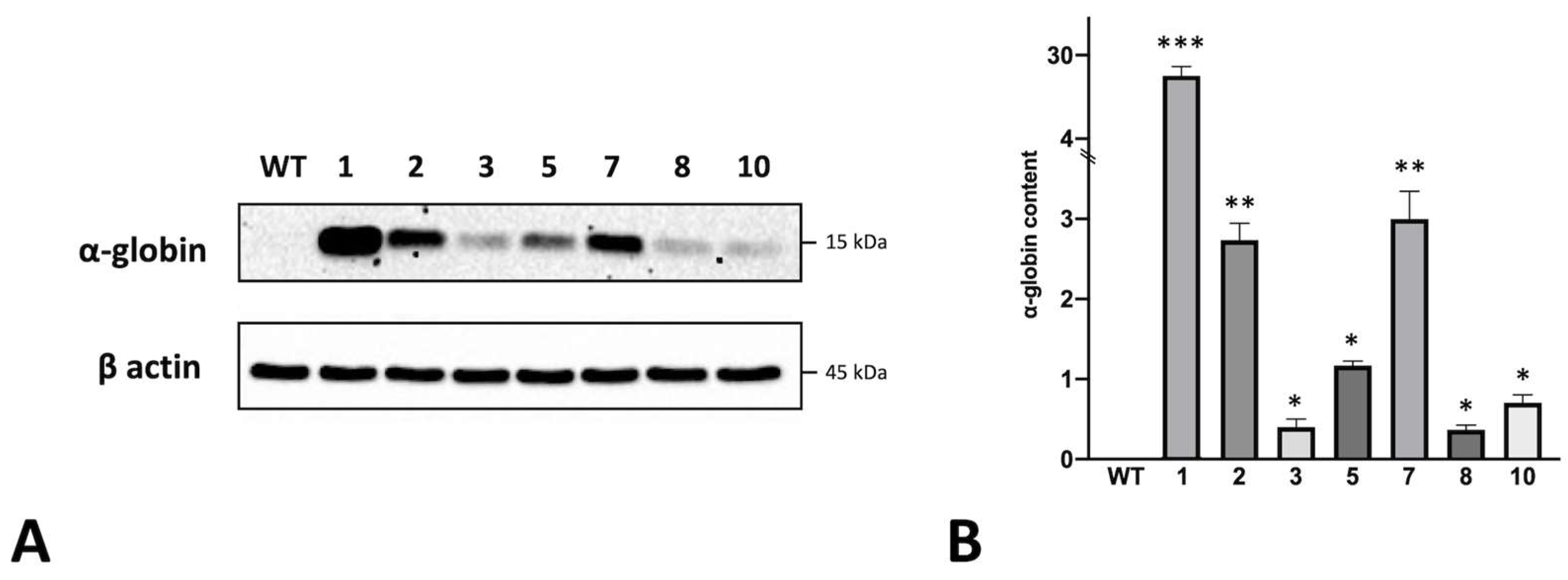
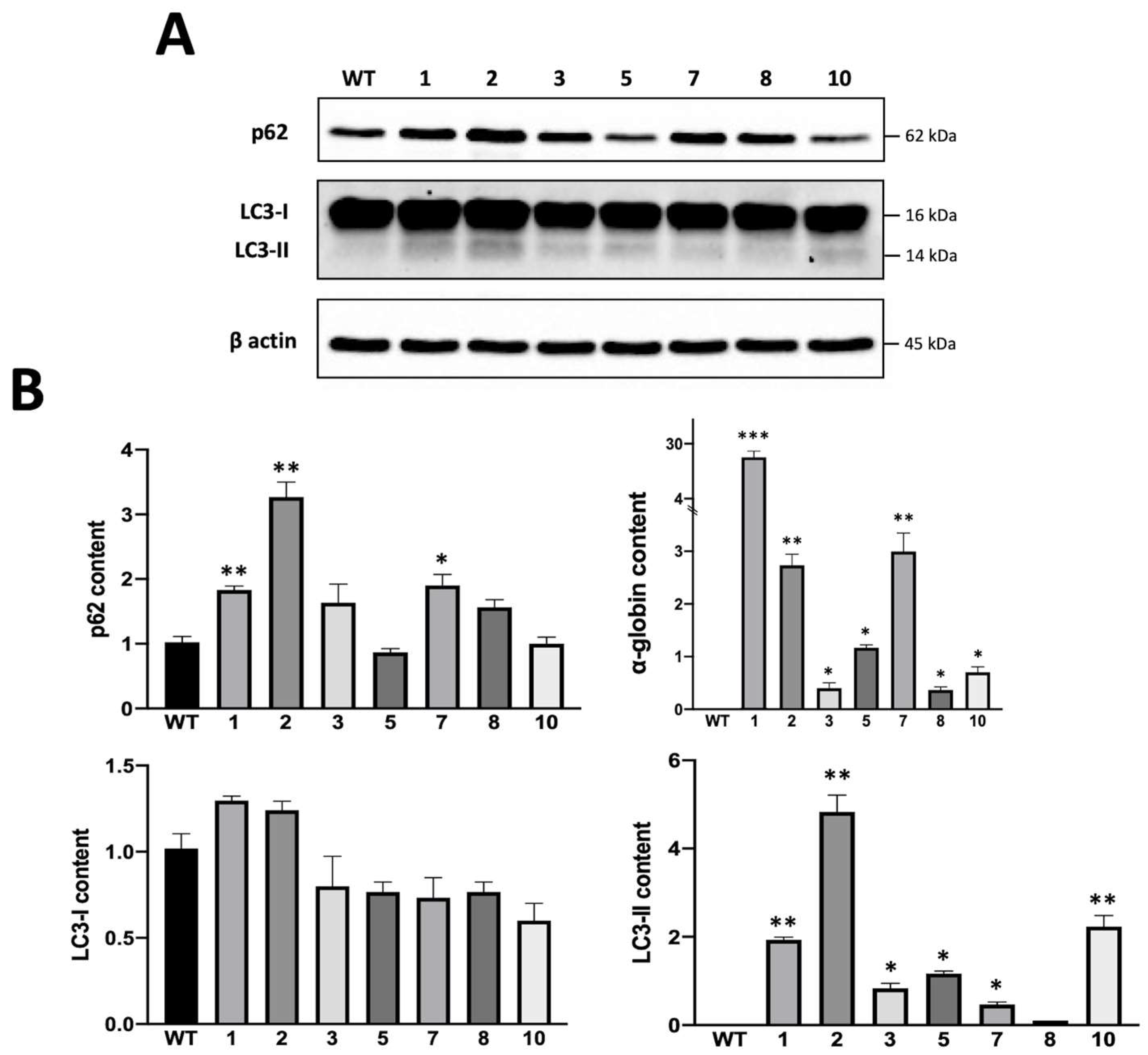
3.2. Preliminary Studies on α-Globin Accumulation in Obtained Clones and Its Relationship with Autophagy
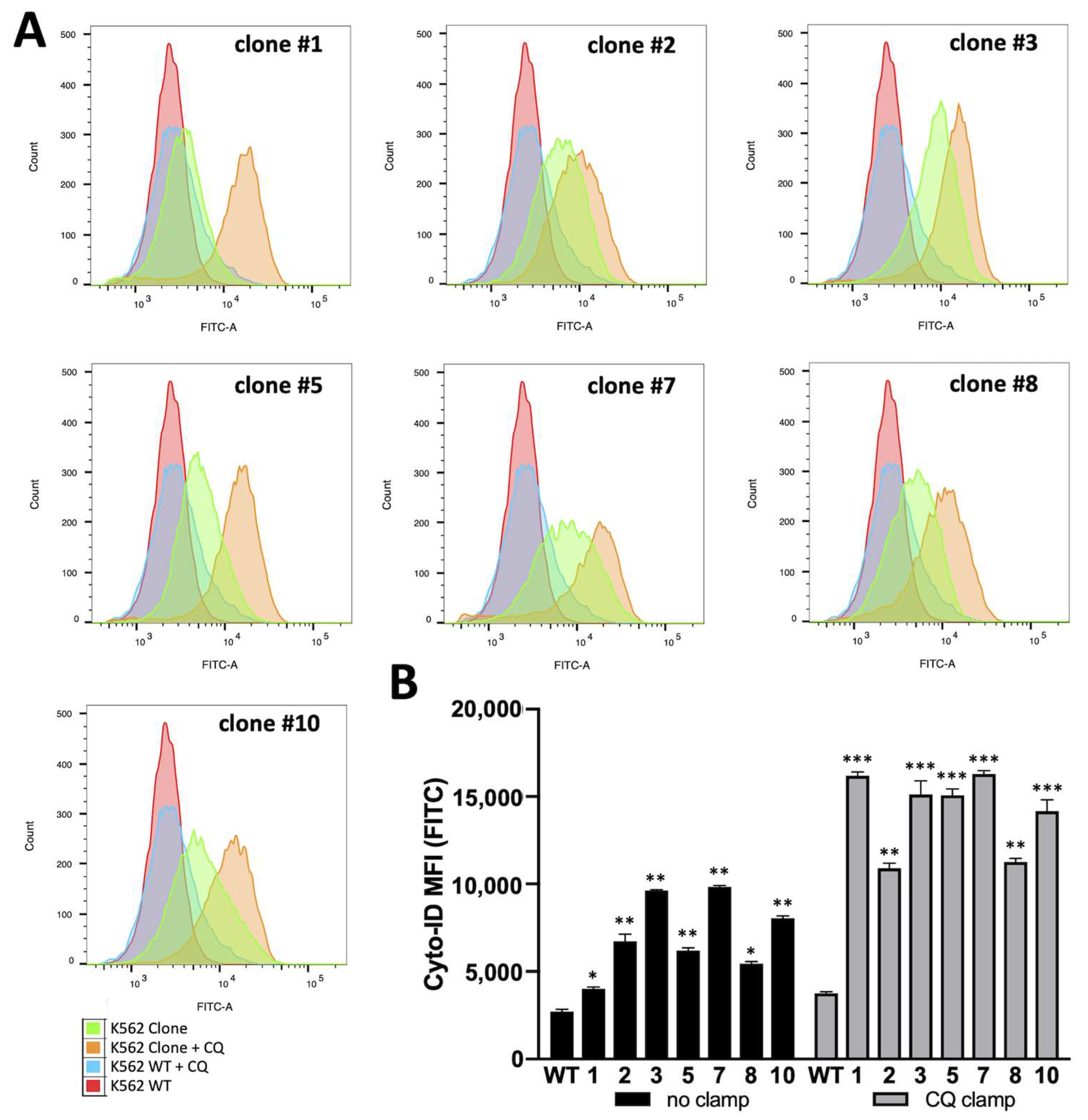
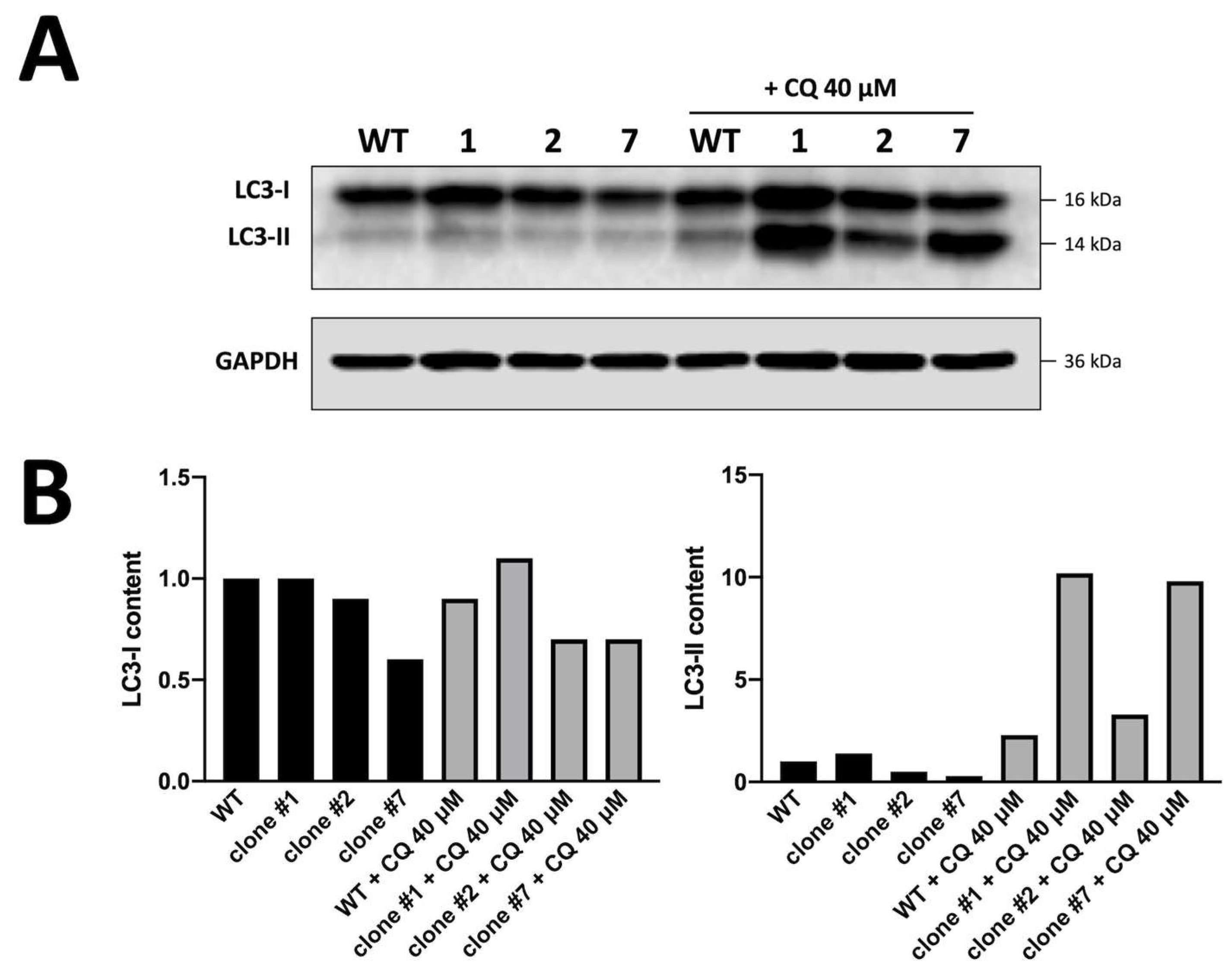
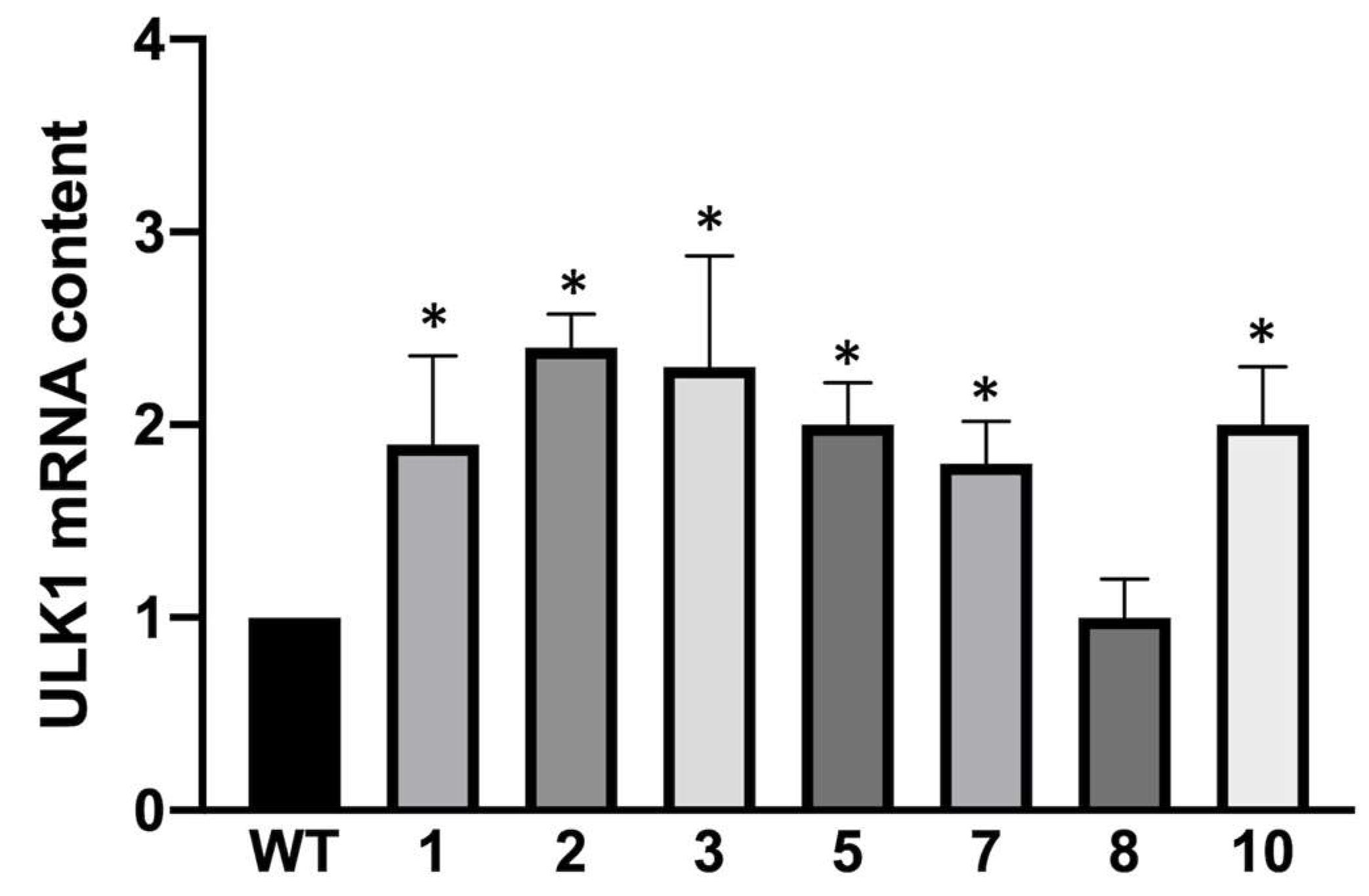
3.3. Biochemical and Molecular Characterization of Autophagy in K562 Cellular Clones Hyper-Expressing α-Globin
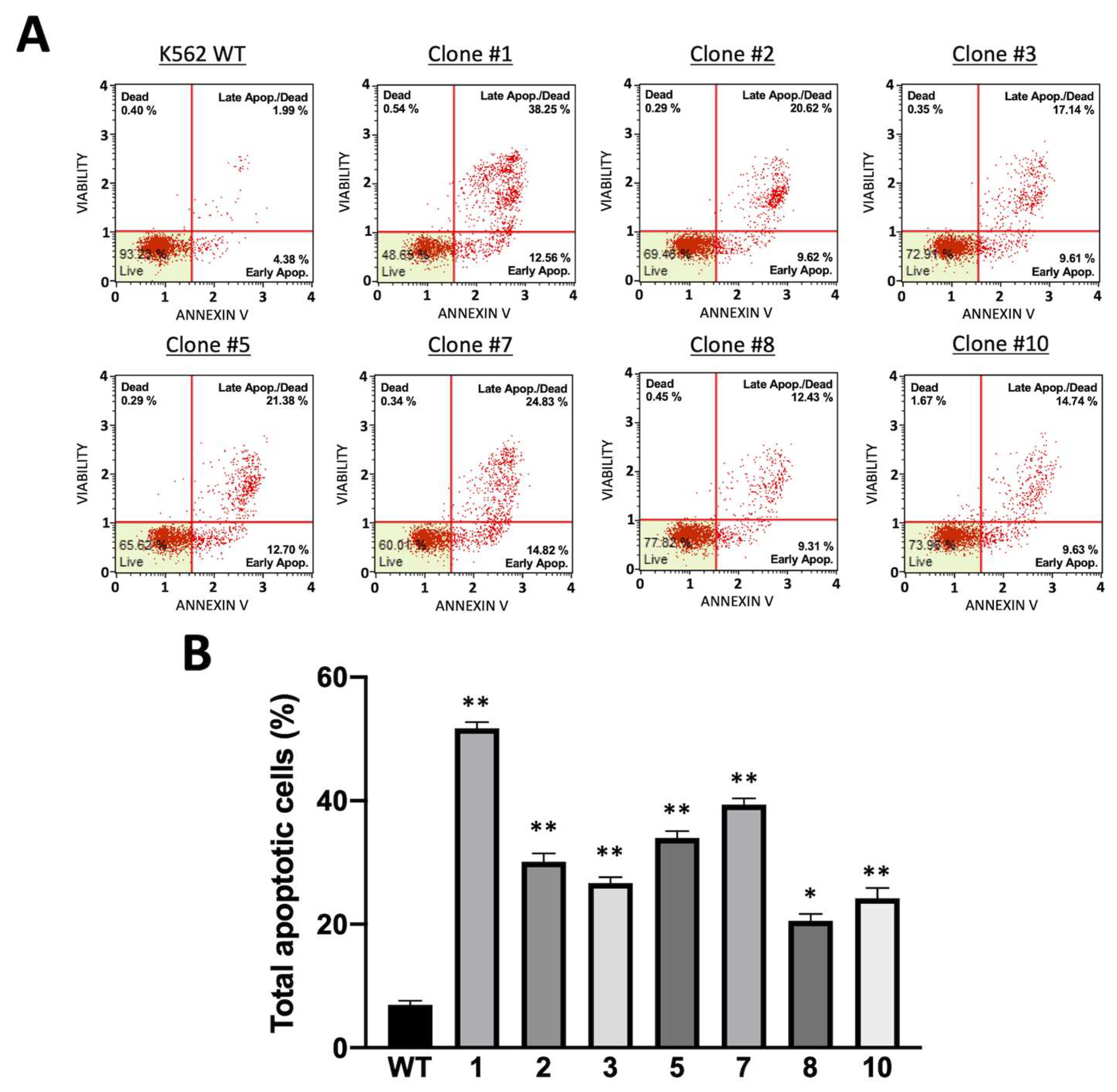
3.4. Phenotypic Characterization of the K562 Cellular Clones Hyper-Expressing α-Globin: Stability of the Plasmid Integration and Tendency to Undergo Apoptosis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weatherall, D.J. Phenotype-genotype relationships in monogenic disease: Lessons from the thalassaemias. Nat. Rev. Genet. 2001, 2, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Origa, R. β-Thalassemia. Genet. Med. 2017, 19, 609–619. [Google Scholar] [CrossRef] [Green Version]
- Fucharoen, S.; Weatherall, D.J. Progress Toward the Control and Management of the Thalassemias. Hematol. Oncol. Clin. N. Am. 2016, 30, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Galanello, R.; Origa, R. β-thalassemia. Orphanet J. Rare Dis. 2010, 5, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cromer, M.K.; Camarena, J.; Martin, R.M.; Lesch, B.J.; Vakulskas, C.A.; Bode, N.M.; Kurgan, G.; Collingwood, M.A.; Rettig, G.R.; Behlke, M.A.; et al. Gene replacement of α-globin with β-globin restores hemoglobin balance in β-thalassemia-derived hematopoietic stem and progenitor cells. Nat. Med. 2021, 27, 677–687. [Google Scholar] [CrossRef]
- Farashi, S.; Vakili, S.; Faramarzi Garous, N.; Ashki, M.; Imanian, H.; Azarkeivan, A.; Najmabadi, H. Copy number variations of six and seven α-globin genes in a family with intermedia and major thalassemia phenotypes. Expert Rev. Hematol. 2015, 8, 693–698. [Google Scholar] [CrossRef]
- Sanchez-Villalobos, M.; Blanquer, M.; Moraleda, J.M.; Salido, E.J.; Perez-Oliva, A.B. New Insights into Pathophysiology of β-Thalassemia. Front. Med. 2022, 9, 880752. [Google Scholar] [CrossRef]
- Rieder, R.F.; James, G.W., 3rd. Imbalance in α and β globin synthesis associated with a hemoglobinopathy. J. Clin. Investig. 1974, 54, 948–956. [Google Scholar] [CrossRef] [Green Version]
- Voon, H.P.; Wardan, H.; Vadolas, J. siRNA-mediated reduction of α-globin results in phenotypic improvements in β-thalassemic cells. Haematologica 2008, 93, 1238–1242. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.Y.; Ren, Z.R.; Zhang, J.Z.; Guo, X.B.; Wang, Q.X.; Wang, S.; Lin, D.; Gong, X.L.; Li, W.; Huang, S.Z.; et al. Restoration of the balanced α/β-globin gene expression in beta654-thalassemia mice using combined RNAi and antisense RNA approach. Hum. Mol. Genet. 2007, 16, 2616–2625. [Google Scholar] [CrossRef] [Green Version]
- Mettananda, S.; Yasara, N.; Fisher, C.A.; Taylor, S.; Gibbons, R.; Higgs, D. Synergistic silencing of α-globin and induction of γ-globin by histone deacetylase inhibitor, vorinostat as a potential therapy for β-thalassaemia. Sci. Rep. 2019, 9, 11649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mettananda, S.; Gibbons, R.J.; Higgs, D.R. α-Globin as a molecular target in the treatment of β-thalassemia. Blood 2015, 125, 3694–3701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechauve, C.; Keith, J.; Khandros, E.; Fowler, S.; Mayberry, K.; Freiwan, A.; Thom, C.S.; Delbini, P.; Romero, E.B.; Zhang, J.; et al. The autophagy-activating kinase ULK1 mediates clearance of free α-globin in β-thalassemia. Sci. Transl. Med. 2019, 11, eaav4881. [Google Scholar] [CrossRef] [PubMed]
- Mettananda, S.; Fisher, C.A.; Hay, D.; Badat, M.; Quek, L.; Clark, K.; Hublitz, P.; Downes, D.; Kerry, J.; Gosden, M.; et al. Editing an α-globin enhancer in primary human hematopoietic stem cells as a treatment for β-thalassemia. Nat. Commun. 2017, 8, 424. [Google Scholar] [CrossRef]
- Pavani, G.; Fabiano, A.; Laurent, M.; Amor, F.; Cantelli, E.; Chalumeau, A.; Maule, G.; Tachtsidi, A.; Concordet, J.P.; Cereseto, A.; et al. Correction of β-thalassemia by CRISPR/Cas9 editing of the α-globin locus in human hematopoietic stem cells. Blood Adv. 2021, 5, 1137–1153. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, K.; Hou, D.X.; Gao, H.; Tang, X.; Kitabayashi, I.; Nishikura, K.; Gachelin, G. Inhibition of expression of a mouse α-globin gene by plasmids that include antisense oligonucleotides. Cell Struct. Funct. 1992, 17, 433–442. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.R.; Ma, J.; Chen, P.; Mo, W.N.; Lin, W.X.; Lai, Y.R. The regulation effect of liposomal transfection of antisense oligonucleotide on the α-globin in patients with severe β-thalassemia. Zhonghua Xueyexue Zazhi 2009, 30, 385–389. [Google Scholar]
- Ma, Y.N.; Chen, M.T.; Wu, Z.K.; Zhao, H.L.; Yu, H.C.; Yu, J.; Zhang, J.W. Emodin can induce K562 cells to erythroid differentiation and improve the expression of globin genes. Mol. Cell. Biochem. 2013, 382, 127–136. [Google Scholar] [CrossRef]
- Papadopoulou, L.C.; Ingendoh-Tsakmakidis, A.; Mpoutoureli, C.N.; Tzikalou, L.D.; Spyridou, E.D.; Gavriilidis, G.I.; Kaiafas, G.C.; Ntaska, A.T.; Vlachaki, E.; Panayotou, G.; et al. Production and Transduction of a Human Recombinant β-Globin Chain into Proerythroid K-562 Cells To Replace Missing Endogenous β-Globin. Mol. Pharm. 2018, 15, 5665–5677. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, Z.; Gao, Q.; Bai, X.; Zhang, C.; Hou, H. Prediction of K562 Cells Functional Inhibitors Based on Machine Learning Approaches. Curr. Pharm. Des. 2019, 25, 4296–4302. [Google Scholar] [CrossRef]
- Alter, B.P.; Goff, S.C. Electrophoretic separation of human embryonic globin demonstrates “α-thalassemia” in human leukemia cell line K562. Biochem. Biophys. Res. Commun. 1980, 94, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Lozzio, B.B.; Lozzio, C.B. Properties of the K562 cell line derived from a patient with chronic myeloid leukemia. Int. J. Cancer 1977, 19, 136. [Google Scholar] [CrossRef]
- Zurlo, M.; Romagnoli, R.; Oliva, P.; Gasparello, J.; Finotti, A.; Gambari, R. Synergistic Effects of A Combined Treatment of Glioblastoma U251 Cells with An Anti-miR-10b-5p Molecule and An AntiCancer Agent Based on 1-(3′,4′,5′-Trimethoxyphenyl)-2-Aryl-1H-Imidazole Scaffold. Int. J. Mol. Sci. 2022, 23, 5991. [Google Scholar] [CrossRef] [PubMed]
- Zurlo, M.; Romagnoli, R.; Oliva, P.; Gasparello, J.; Finotti, A.; Gambari, R. Synergistic effects of the combined treatment of U251 and T98G glioma cells with an anti-tubulin tetrahydrothieno [2,3-c]pyridine derivative and a peptide nucleic acid targeting miR-221-3p. Int. J. Oncol. 2021, 59, 61. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Cosenza, L.C.; Zurlo, M.; Gasparello, J.; Papi, C.; D’Aversa, E.; Breveglieri, G.; Lampronti, I.; Finotti, A.; Borgatti, M.; et al. Expression of γ-globin genes in β-thalassemia patients treated with sirolimus: Results from a pilot clinical trial (Sirthalaclin). Ther. Adv. Hematol. 2022, 13, 20406207221100648. [Google Scholar] [CrossRef]
- Murugan, S.; Amaravadi, R.K. Methods for Studying Autophagy Within the Tumor Microenvironment. Adv. Exp. Med. Biol. 2016, 899, 145–166. [Google Scholar] [CrossRef] [Green Version]
- Cosenza, L.C.; Gasparello, J.; Romanini, N.; Zurlo, M.; Zuccato, C.; Gambari, R.; Finotti, A. Efficient CRISPR-Cas9-based genome editing of β-globin gene on erythroid cells from homozygous β039-thalassemia patients. Mol. Ther. Methods Clin. Dev. 2021, 21, 507–523. [Google Scholar] [CrossRef]
- Gasparello, J.; Papi, C.; Zurlo, M.; Cosenza, L.C.; Breveglieri, G.; Zuccato, C.; Gambari, R.; Finotti, A. Teaching during COVID-19 pandemic in practical laboratory classes of applied biochemistry and pharmacology: A validated fast and simple protocol for detection of SARS-CoV-2 Spike sequences. PLoS ONE 2022, 17, e0266419. [Google Scholar] [CrossRef]
- Zuccato, C.; Cosenza, L.C.; Zurlo, M.; Lampronti, I.; Borgatti, M.; Scapoli, C.; Gambari, R.; Finotti, A. Treatment of Erythroid Precursor Cells from β-Thalassemia Patients with Cinchona Alkaloids: Induction of Fetal Hemoglobin Production. Int. J. Mol. Sci. 2021, 22, 13433. [Google Scholar] [CrossRef]
- Delgado-Cañedo, A.; Chies, J.A.; Nardi, N.B. Induction of fetal haemoglobin expression in erythroid cells--a model based on iron availability signalling. Med. Hypotheses 2005, 65, 932–936. [Google Scholar] [CrossRef]
- Kurita, R.; Suda, N.; Sudo, K.; Miharada, K.; Hiroyama, T.; Miyoshi, H.; Tani, K.; Nakamura, Y. Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PLoS ONE 2013, 8, e59890. [Google Scholar] [CrossRef] [Green Version]
- Vinjamur, D.S.; Bauer, D.E. Growing and Genetically Manipulating Human Umbilical Cord Blood-Derived Erythroid Progenitor (HUDEP) Cell Lines. Methods Mol. Biol. 2018, 1698, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Perera, S.; Allen, A.; Silva, I.; Hapugoda, M.; Wickramarathne, M.N.; Wijesiriwardena, I.; Allen, S.; Rees, D.; Efremov, D.G.; Fisher, C.A.; et al. Genotype-phenotype association analysis identifies the role of α globin genes in modulating disease severity of β thalassaemia intermedia in Sri Lanka. Sci. Rep. 2019, 9, 10116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandros, E.; Thom, C.S.; D’Souza, J.; Weiss, M.J. Integrated protein quality-control pathways regulate free α-globin in murine β-thalassemia. Blood 2012, 119, 5265–5275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Wang, H.; Zhang, D.; Luo, W.; Liu, R.; Xu, D.; Diao, L.; Liao, L.; Liu, Z. Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat. Commun. 2018, 9, 3492. [Google Scholar] [CrossRef]
- Hu, Z.; Sankar, D.S.; Vu, B.; Leytens, A.; Vionnet, C.; Wu, W.; Stumpe, M.; Martínez-Martínez, E.; Stork, B.; Dengjel, J. ULK1 phosphorylation of striatin activates protein phosphatase 2A and autophagy. Cell Rep. 2021, 36, 109762. [Google Scholar] [CrossRef]
- Poole, L.P.; Bock-Hughes, A.; Berardi, D.E.; Macleod, K.F. ULK1 promotes mitophagy via phosphorylation and stabilization of BNIP3. Sci. Rep. 2021, 11, 20526. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Fan, W.; Zhong, Q. Regulation of Beclin 1 in autophagy. Autophagy 2009, 5, 713–716. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [Green Version]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Nah, J.; Oka, S.I.; Mukai, R.; Monden, Y.; Maejima, Y.; Ikeda, Y.; Sciarretta, S.; Liu, T.; Li, H. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J. Clin. Investig. 2019, 129, 802–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers and Probes | Sequences |
|---|---|
| forward primer α-globin | 5′-CGACAAGACCAACGTCAAGG-3′ |
| reverse primer α-globin | 5′-GGTCTTGGTGGTGGGGAAG-3′ |
| probe α-globin | 5′-HEX-ACATCCTCTCCAGGGCCTCCG-BFQ-3′ |
| forward primer ULK1 | 5′-CTACCTGGTTATGGAGTACTGC-3′ |
| reverse primer ULK1 | 5′-GGAAGAGCCTGATGGTGTC-3′ |
| probe ULK1 | 5′-FAM-CGACTACCT/ZEN/GCACGCCATGC-BFQ-3′ |
| forward primer RPL13A | 5′-GGCAATTTCTACAGAAACAAGTTG-3′ |
| reverse primer RPL13A | 5′-GTTTTGTGGGGCAGCATACC-3′ |
| probe RPL13A | 5′-HEX-CGCACGGTCCGCCAGAAGAT-BFQ-3′ |
| forward primer ACTB | 5′-ACAGAGCCTCGCCTTTG-3′ |
| reverse primer ACTB | 5′-ACGATGGAGGGGAAGACG-3′ |
| probe ACTB | 5′-Cy5-CCTTGCACATGCCGGAGCC-BRQ-3′ |
| forward primer GAPDH | 5′-ACATCGCTCAGACACCATG-3′ |
| reverse primer GAPDH | 5′-TGTAGTTGAGGTCAATGAAGGG-3′ |
| probe GAPDH | 5′-FAM-AAGGTCGGAGTCAACGGATTTGGTC-BFQ-3′ |
| Target | Primary Antibody | Cat. n. | Secondary Antibody | Cat. n. |
|---|---|---|---|---|
| α-globin | Mouse anti-hemoglobin α (D-4) (Santa Cruz Biotechnology, Dallas, TX, USA) | sc-514378 | Goat anti-mouse IgG HRP (Thermo Fisher, Waltham, MA, USA) | 32430 |
| p62 | Rabbit anti-p62/SQSTM1 (Sigma-Aldrich, St. Louis, MO, USA) | P0067 | Mouse anti-rabbit IgG HRP (Cell Signaling Technology, Danvers, MA, USA | 7074 |
| LC3 | Rabbit anti-LC3B (Sigma-Aldrich, St. Louis, MO, USA) | L7543 | Mouse anti-rabbit IgG HRP (Cell Signaling Technology, Danvers, MA, USA) | 7074 |
| β-actin | Rabbit anti-β-actin (Cell Signaling Technology, Danvers, MA, USA) | 4967 | Mouse anti-rabbit IgG HRP (Cell Signaling Technology, Danvers, MA, USA) | 7074 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zurlo, M.; Gasparello, J.; Cosenza, L.C.; Breveglieri, G.; Papi, C.; Zuccato, C.; Gambari, R.; Finotti, A. Production and Characterization of K562 Cellular Clones Hyper-Expressing the Gene Encoding α-Globin: Preliminary Analysis of Biomarkers Associated with Autophagy. Genes 2023, 14, 556. https://doi.org/10.3390/genes14030556
Zurlo M, Gasparello J, Cosenza LC, Breveglieri G, Papi C, Zuccato C, Gambari R, Finotti A. Production and Characterization of K562 Cellular Clones Hyper-Expressing the Gene Encoding α-Globin: Preliminary Analysis of Biomarkers Associated with Autophagy. Genes. 2023; 14(3):556. https://doi.org/10.3390/genes14030556
Chicago/Turabian StyleZurlo, Matteo, Jessica Gasparello, Lucia Carmela Cosenza, Giulia Breveglieri, Chiara Papi, Cristina Zuccato, Roberto Gambari, and Alessia Finotti. 2023. "Production and Characterization of K562 Cellular Clones Hyper-Expressing the Gene Encoding α-Globin: Preliminary Analysis of Biomarkers Associated with Autophagy" Genes 14, no. 3: 556. https://doi.org/10.3390/genes14030556