

Pathogenicity Analysis of a Novel Variant in GTPBP3 Causing Mitochondrial Disease and Systematic Literature Review

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Clinical Characteristics of the Patient
2.2. Molecular Genetic Analysis
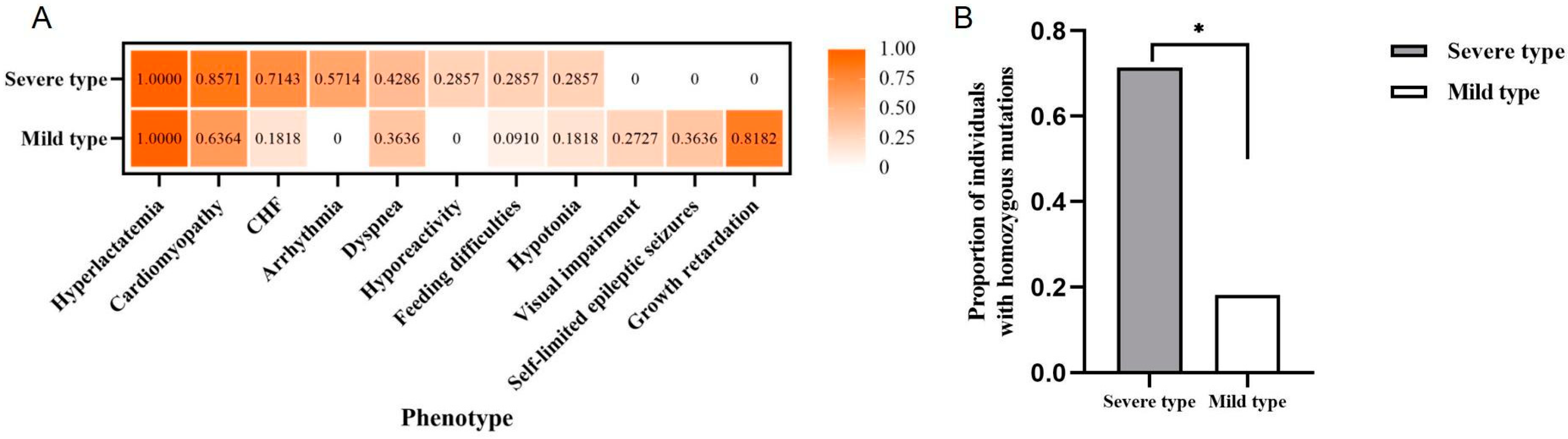
2.3. Systematic Analysis on Clinical Phenotypes of Individuals with GTPBP3 Variants
2.4. Variant Spectrum and Distribution of GTPBP3 in the Severe and Mild Type
3. Discussion
4. Materials and Methods
4.1. Patient and Clinical Evaluation
4.2. Trio Whole-Exome Sequencing and Sanger Sequencing
4.3. Phenotypic Analysis of Individuals with GTPBP3 Variants
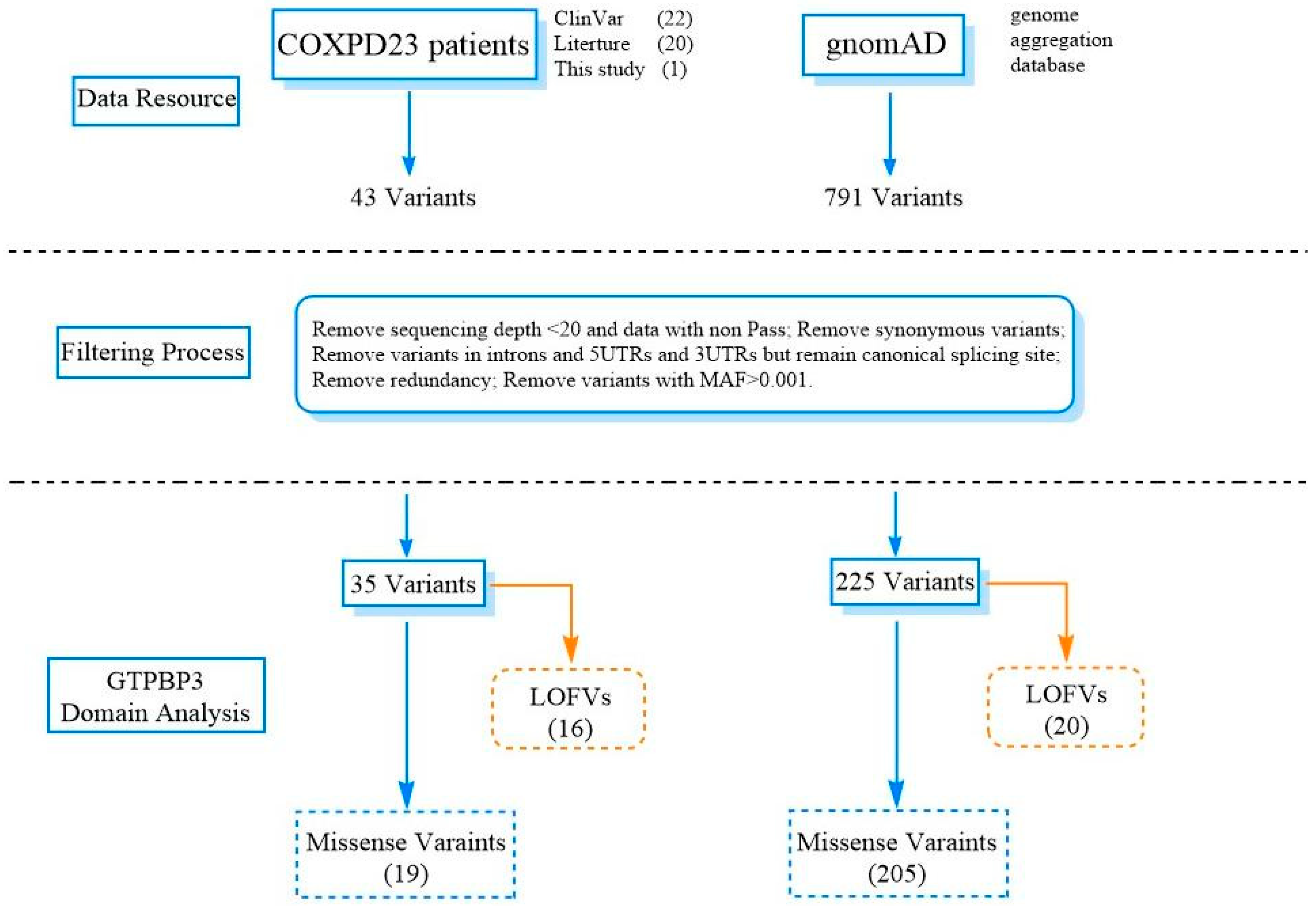
4.4. Comprehensive Analysis of GTPBP3 Variants
4.5. Distribution of Variants in GTPBP3 Domains
4.6. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Craven, L.; Alston, C.L.; Taylor, R.W.; Turnbull, D.M. Recent Advances in Mitochondrial Disease. Annu. Rev. Genom. Hum. Genet. 2017, 18, 257–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, G.X.; Zhang, Y.; Wang, Q.Q.; Li, Q.R.; Xu, H.; Wang, E.D.; Zhou, X.L. The human tRNA taurine modification enzyme GTPBP3 is an active GTPase linked to mitochondrial diseases. Nucleic Acids Res. 2021, 49, 2816–2834. [Google Scholar] [CrossRef] [PubMed]
- Kopajtich, R.; Nicholls, T.J.; Rorbach, J.; Metodiev, M.D.; Freisinger, P.; Mandel, H.; Vanlander, A.; Ghezzi, D.; Carrozzo, R.; Taylor, R.W.; et al. Mutations in GTPBP3 cause a mitochondrial translation defect associated with hypertrophic cardiomyopathy, lactic acidosis, and encephalopathy. Am. J. Hum. Genet. 2014, 95, 708–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.M.; Liu, Z.M.; Cao, B.; Zhang, V.W.; He, Y.D.; Jia, Z.J.; Xi, H.; Liu, J.; Fang, F.; Wang, H. Novel Mutations in the GTPBP3 Gene for Mitochondrial Disease and Characteristics of Related Phenotypic Spectrum: The First Three Cases from China. Front. Genet. 2021, 12, 611226. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.F.; Hou, C.C.; Yang, W.X. Nuclear factors: Roles related to mitochondrial deafness. Gene 2013, 520, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Villarroya, M.; Prado, S.; Esteve, J.M.; Soriano, M.A.; Aguado, C.; Perez-Martinez, D.; Martinez-Ferrandis, J.I.; Yim, L.; Victor, V.M.; Cebolla, E.; et al. Characterization of human GTPBP3, a GTP-binding protein involved in mitochondrial tRNA modification. Mol. Cell. Biol. 2008, 28, 7514–7531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asano, K.; Suzuki, T.; Saito, A.; Wei, F.Y.; Ikeuchi, Y.; Numata, T.; Tanaka, R.; Yamane, Y.; Yamamoto, T.; Goto, T.; et al. Metabolic and chemical regulation of tRNA modification associated with taurine deficiency and human disease. Nucleic Acids Res. 2018, 46, 1565–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Gao, Y.; Ning, Z.; Lu, Y.; Zhang, X.; Liu, J.; Xie, B.; Xue, Z.; Wang, X.; Yuan, K.; et al. PGG.SNV: Understanding the evolutionary and medical implications of human single nucleotide variations in diverse populations. Genome Biol. 2019, 20, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Zuo, C.; Liao, S.; Long, Y.; Wang, Y. Abnormal development pattern of the amygdala and hippocampus from childhood to adulthood with autism. J. Clin. Neurosci. 2020, 78, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Fu, Y.Q. Mitochondrial diseases associated with mutations in GTPBP3: A case report and literature review. J. Clin. Pediatr. 2021, 39, 818–821. [Google Scholar] [CrossRef]
- Zhao, X.X.; Wu, X.Y.; Xu, L.J.; Wang, D.; Jiang, W.; Song, L.; Kang, L.M. Mitochondrial Cardiomyopathy Caused by Nuclear Gene Mutation: A Case Report and Literature Review. Mol. Cardiol. China 2021, 21, 4387–4389. [Google Scholar] [CrossRef]
- Elmas, M.; Yildiz, H.; Erdogan, M.; Gogus, B.; Avci, K.; Solak, M. Comparison of clinical parameters with whole exome sequencing analysis results of autosomal recessive patients; a center experience. Mol. Biol. Rep. 2019, 46, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhang, Z.; Chen, C.; Yao, S.; Yang, Q.; Li, F.; He, X.; Ai, C.; Wang, M.; Guan, M.X. Deletion of Gtpbp3 in zebrafish revealed the hypertrophic cardiomyopathy manifested by aberrant mitochondrial tRNA metabolism. Nucleic Acids Res. 2019, 47, 5341–5355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Zamora, A.; Meseguer, S.; Esteve, J.M.; Villarroya, M.; Aguado, C.; Enriquez, J.A.; Knecht, E.; Armengod, M.E. Defective Expression of the Mitochondrial-tRNA Modifying Enzyme GTPBP3 Triggers AMPK-Mediated Adaptive Responses Involving Complex I Assembly Factors, Uncoupling Protein 2, and the Mitochondrial Pyruvate Carrier. PLoS ONE 2015, 10, e0144273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutoual, R.; Meseguer, S.; Villarroya, M.; Martin-Hernandez, E.; Errami, M.; Martin, M.A.; Casado, M.; Armengod, M.E. Defects in the mitochondrial-tRNA modification enzymes MTO1 and GTPBP3 promote different metabolic reprogramming through a HIF-PPARgamma-UCP2-AMPK axis. Sci. Rep. 2018, 8, 1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Zhang, C.; Yuan, L.; Ling, Y.; Wang, X.; Liu, C.; Pan, Y.; Zhang, X.; Ma, X.; Wang, Y.; et al. PGG.Han: The Han Chinese genome database and analysis platform. Nucleic Acids Res. 2020, 48, D971–D976. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Zhang, L.; Liu, W.; Jiang, Y.; Chen, X.; Lan, X.; Li, G.; Hang, Q.; Wang, J.; Gusella, J.F.; et al. De novo variants in the Helicase-C domain of CHD8 are associated with severe phenotypes including autism, language disability and overgrowth. Hum. Genet. 2020, 139, 499–512. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| gDNA (GRCh38) | Ex. | Variant (NM_032620.4) | Mut. Type | Inheritance | Allele Freq. | SIFT | Polyphen-2 | CADD | REVEL | Variants (In Trans) | AA Change (In Trans) | Related-Domain | Resources |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| chr19-17337613 | Ex.1 | c.2T>A, p.Met1Lys | Missense | UNK | n.d | D | D | D | 0.342 | T | ClinVar:488854 | ||
| chr19-17337619 | Ex.1 | c.8G>T, p.Arg3Leu | Missense | UNK | 0.0006418 | D | D | D | 0.096 | c.934_957del | p.Gly312_Val319del | T | Kopajtich et al. (2014) [3] |
| chr19-17337628 | Ex.1 | c.17G>A, p.Trp6Ter | Nonsense | UNK | 0.00001314 | T/N/C1/G/C2 | ClinVar:1405745 | ||||||
| chr19-17337643-17337644 | Ex.1 | c.32_33delinsGTG, p.Gln11Argfs*98 | Frameshift | UNK | n.d | Homo | Homo | T/N/C1/G/C2 | Kopajtich et al. (2014) [3] | ||||
| chr19-17338109 | Ex.2 | c.155G>A, p.Arg52Gln | Missense | UNK | 0.00001604 | D | D | D | 0.635 | N | ClinVar:372869 | ||
| chr19-17338563 | Ex.4 | c.413C>T, p.Ala138Val | Missense | Paternal | 0.00001196 | D | D | D | 0.580 | c.509_510del | p.Glu170Glyfs*42 | N | Yan et al. (2021) [4] |
| chr19-17338574 | Ex.4 | c.424G>A, p.Glu142Lys | Missense | Maternal | n.d | D | D | D | 0.819 | Homo/ c.689A>C | Homo/ p.Gln230Pro | N | Kopajtich et al. (2014) [3]; Yan et al. (2021) [4] |
| chr19-17338626 | Ex.4 | c.476A>T, p.Glu159Val | Missense | UNK | n.d | D | D | D | 0.893 | c.964G>C | p.Ala322Pro | N | Kopajtich et al. (2014) [3] |
| chr19-17338634 | Ex.4 | c.484G>C, p.Ala162Pro | Missense | Maternal | n.d | D | D | D | 0.371 | c.673G>A and c.964G>C | p.Glu225Lys and p.Ala322Pro | C1 | Kopajtich et al. (2014) [3] |
| chr19-17338659-17338660 | Ex.4 | c.509_510del, p.Glu170Glyfs*42 | Frameshift | Maternal | 0.000006569 | c.413C>T | p.Ala138Val | C1/G/C2 | Yan et al. (2021) [4] | ||||
| chr19-17338662 | Ex.4 | c.512del, p.Ala171Glyfs*82 | Frameshift | UNK | n.d | C1/G/C2 | ClinVar:1389465 | ||||||
| chr19-17338667 | Ex.4 | c.517C>T, p.Arg173Trp | Missense | Paternal | 0.000004009 | D | D | D | 0.300 | c.643G>T | p.Glu215Ter | C1 | ClinVar:488526 |
| chr19-17338671-17338677 | Ex.4 | c.521_527del, p.Arg174Profs*77 | Frameshift | UNK | n.d | 0.256 | C1/G/C2 | ClinVar:1075628 | |||||
| chr19-17338694 | Ex.4 | c.544G>T, p.Gly182X | Missense | Paternal | n.d | - | - | D | - | c.689A>C | p.Gln230Pro | C1 | Yan et al. (2021) [4] |
| chr19-17339005 | Ex.5 | c.643G>T, p.Glu215Ter | Nonsense | Maternal | n.d | c.517C>T | p.Arg173Trp | C1/G/C2 | ClinVar:488527 | ||||
| chr19-17339131 | Ex.6 | c.673G>A, p.Glu225Lys | Missense | Paternal | 0.00001601 | T | T | T | 0.018 | c.484G>C | p.Ala162Pro | C1 | Kopajtich et al. (2014) [3] |
| chr19-17339147 | Ex.6 | c.689A>C, p.Gln230Pro | Missense | Maternal | 0.0001089 | T | D | T | 0.075 | c.424G>A/c.544G>T /c.1073delG/c.1102dupC /c.1280delC | p.Glu142Lys/p.Gly182X /p.Gly358Glufs*16/p.Arg368Profs*22 /p.Pro427Argfs*3 | C1 | Yan et al. (2021) [4]; Yang et al. (2021) [10]; Zhao et al. (2021) [11]; Our patient |
| chr19-17339206 | Ex.6 | c.748del, p.Val250Cysfs*3 | Frameshift | UNK | n.d | G/C2 | ClinVar:1324519 | ||||||
| chr19-17339228 | Ex.6 | c.770C>A, p.Pro257His | Missense | UNK | n.d | T | D | D | 0.715 | Homo | Homo | G | Kopajtich et al. (2014) [3] |
| chr19-17339461 | Ex.7 | c.836C>T, p.Pro279Leu | Missense | UNK | 0.00008873 | D | D | D | 0.323 | Homo | Homo | G | Eimas et al. (2019) [12] |
| chr19-17339490 | Ex.7 | c.865G>T, p.Glu289Ter | Nonsense | UNK | n.d | G/C2 | ClinVar:642459 | ||||||
| chr19-17339550 | Ex.7 | c.925dup, p.Glu309Glyfs*22 | Frameshift | UNK | n.d | G/C2 | ClinVar:1452744 | ||||||
| chr19-17339559-17339582 | Ex.7 | c.934_957del, p.Gly312_Val319del | Non-frameshift deletion | UNK | n.d | - | - | - | - | c.8G>T | p.Arg3Leu | G | Kopajtich et al. (2014) [3] |
| chr19-17339589 | Ex.7 | c.964G>C, p.Ala322Pro | Missense | Paternal | 0.0001104(total) | D | D | D | 0.635 | c.484G>C | p.Ala162Pro | G | Kopajtich et al. (2014) [3] |
| chr19-17341078 | Ex.8 | c.1009G>C, p.Asp337His | Missense | UNK | n.d | D | D | D | 0.796 | Homo | Homo | G | Kopajtich et al. (2014) [3] |
| chr19-17341118 | Ex.8 | c.1049del, p.Leu350Argfs*5 | Frameshift | UNK | n.d | G/C2 | ClinVar:280249 | ||||||
| chr19-17341142 | Ex.8 | c.1073del, p.Gly358Glufs*16 | Frameshift | Paternal | n.d | c.689A>C | p.Gln230Pro | G/C2 | Yang et al. (2021) [10] | ||||
| chr19-17341171 | Ex.8 | c.1102dup, p.Arg368Profs*22 | Frameshift | Maternal | n.d | c.689A>C | p.Gln230Pro | G/C2 | Our Patient | ||||
| chr19-17341181 | Ex.8 | c.1112T>C, p.Leu371Pro | Missense | Paternal | 0.000004009 | T | D | D | 0.314 | c.440C>T | p.Ala147Val | G | ClinVar:488528 |
| chr19-17341504 | Ex.9 | c.1280del, p.Pro427Argfs*3 | Frameshift | Maternal | n.d | c.689A>C | p.Gln230Pro | C2 | Zhao et.al. (2021) [11] | ||||
| chr19-17341515 | Ex.9 | c.1291dup, p.Pro430Argfs*86 | Frameshift | UNK | n.d | C2 | ClinVar:180614 | ||||||
| chr19-17341599 | Ex.9 | c.1375G>A, p.Glu459Lys | Missense | UNK | n.d | D | D | D | 0.739 | c.1291dupC | p.Pro430Argfs*86 | C2 | Kopajtich et al. (2014) [3] |
| chr19-17341663 | Ex.9 | c.1439T>A, p.Ile480Asn | Missense | UNK | n.d | D | D | D | 0.515 | C2 | ClinVar:800909 |
| Patient No. | SP202010 | #Case | #Case | #2 a | #3 b |
| Our Patient | Zhao XX et al. (2021) [11] | Yang Q et al. (2021) [10] | Yan HM et al. (2021) [4] | Yan HM et al. (2021) [4] | |
| Variants | c.1102dupC, p.Arg368Profs*22(mat.) c.689A>C, p.Gln230Pro(pat.) m.8108A>G (100%) | c.1280delC, p.Pro427Argfs*3(mat.) c.689A>C, p.Gln230Pro(pat.) | c.689A>C, p.Gln230Pro (mat.) c.1073delG, p.Gly358Glufs*16(pat.) | c.689A>C, p.Gln230Pro(mat.) c.544G>T, p.Gly182*(pat.) | c.424G>A, p.Glu142Lys(mat.) c.689A>C, p.Gln230Pro(pat.) |
| Gender | M | F | F | F | F |
| AO | 3 days | 9 years | 3 years | 1 year | 1 year |
| Movement | Could walk but not run or jump | Could walk but had weakness in both lower limbs | Could walk slowly | Could not walk | Could walk or run but easily fell down |
| Language | Speak 5–6 words and unclear pronunciation | UNK | Speak a few words but no complete sentences | Could not speak | Speak a few words but no complete sentences |
| Other phenotypes | Intellectual disability and motor developmental delay; self-limited epileptic seizures; abnormal visual development; short stature | Heart failure; intellectual developmental delay; abdominal pain; abdominal distention | Respiratory failure; myocardial damage; stroke-like syndrome, global developmental delay | Developmental delay; hypotonia | Developmental delay; intellectual disability; fatigability |
| TTE | HCM (LVH) | HCM (LVH) | HCM (LVH) | UNK | HCM (LVH) |
| Brain MRI | Abnormal signal of bilateral thalamus | UNK | Abnormal signal of the bilateral cortical spinal tract | bilateral lesions in the midbrain, thalamus, and dentate body of the cerebellum | Bilateral lesions in the brain stem, thalamus, and dentate body of the cerebellum |
| Plasma | Lactate: 3–11.6 mmol/L↑ Normal amino acid and acylcarnitine profiles | Lactate: 5.53 mmol/L↑ N-terminal pro-BNP: 1603.3 pg/mL↑ | Lactate: 29 mmol/L↑ CK-MB: 15.92μg/L, Pyruvate 599μml/L β-hydroxybutyrate Acid 0. 71 mmol/L, blood ammonia: 45.1μmol/L | Lactate: 7.7∼14 mmol/L↑ CK-MB: 32 U/L, Hyperalanine: 256.75 mmol/L normal acylcarnitine profiles | Lactate: 4.26–16 mmol/L↑ |
| MS (Urine) | Increased levels of lactic acid, 3-hydroxybutyric acid, 3-methylglutaconic acid, 3-hydroxyglutaric acid, and hippuric acid | UNK | UNK | Increased level of Lactic acid, | Normal organic acid profile |
| EEG/ECG | High-amplitude 2–4 Hz sharp slow wave paroxysms mainly in the right occipital region, sometimes spreading to both frontal and occipital regions | UNK | Bilateral occipital rhythm dominated by 5–7 Hz θ rhythm, no paroxysmal EEG | UNK | Normal |
| Outcomes; cause of death | Alive after 10 years | Alive after 17 years | Alive after 3 years | Alive after 3 years | Alive after 3 years |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Ouyang, Q.; Xiang, J.; Li, H.; Lv, H.; An, Y. Pathogenicity Analysis of a Novel Variant in GTPBP3 Causing Mitochondrial Disease and Systematic Literature Review. Genes 2023, 14, 552. https://doi.org/10.3390/genes14030552
Zhang Q, Ouyang Q, Xiang J, Li H, Lv H, An Y. Pathogenicity Analysis of a Novel Variant in GTPBP3 Causing Mitochondrial Disease and Systematic Literature Review. Genes. 2023; 14(3):552. https://doi.org/10.3390/genes14030552
Chicago/Turabian StyleZhang, Qin, Qianqian Ouyang, Jingjing Xiang, Hong Li, Haitao Lv, and Yu An. 2023. "Pathogenicity Analysis of a Novel Variant in GTPBP3 Causing Mitochondrial Disease and Systematic Literature Review" Genes 14, no. 3: 552. https://doi.org/10.3390/genes14030552