
TCF7L1 Regulates LGR5 Expression in Colorectal Cancer Cells
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Analysis of the Cancer Genome Atlas (TCGA) Datasets
2.2. Transcript Analysis in Patient-Matched Samples
2.3. Cell Culture
2.4. Plasmids
2.5. Stable Cell Lines
2.6. Reverse Transcription and Real Time PCR (RT-qPCR)
2.7. Western Blot
2.8. Chromatin Immunoprecipitation (ChIP)
2.9. Luciferase Reporter Assays
2.10. DNA Binding Assay
2.11. Spheroid Formation Assays
2.12. CRISPR Activation/Interference (CRISPRa/i)
2.13. Statistics
3. Results
3.1. TCF7L1 Expression Is Downregulated and LGR5 Expression Is Upregulated in Patient Tumor Samples
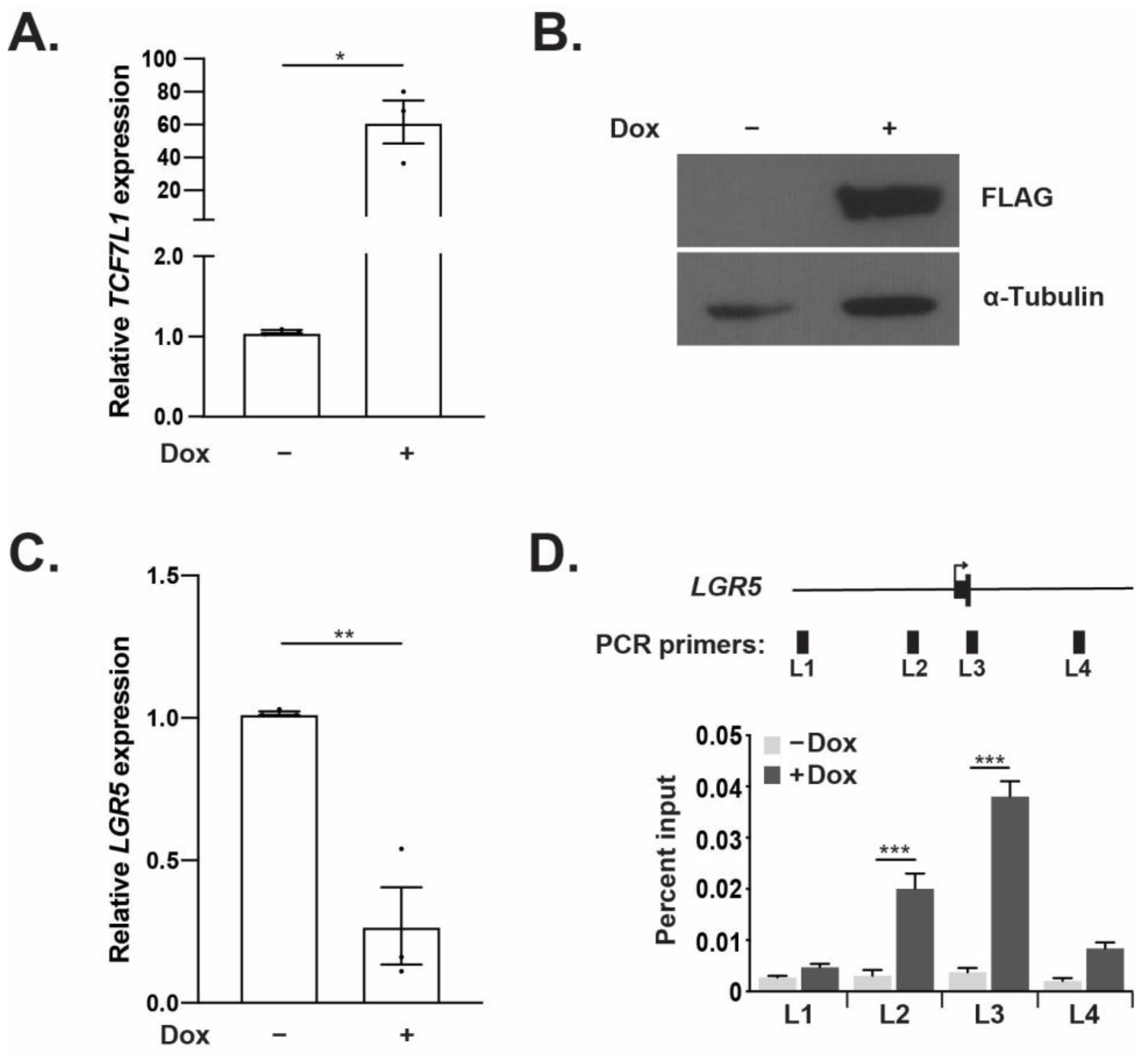
3.2. TCF7L1 Represses LGR5 Expression and Directly Binds the LGR5 Promoter Region
3.3. TCF7L1 Occupancy Demarcates a WRE at the LGR5 Locus That Requires a Single TBE for Full Activity
3.4. TCF7L1 Reduces Spheroid Formation Efficiency of CRC Cell Lines
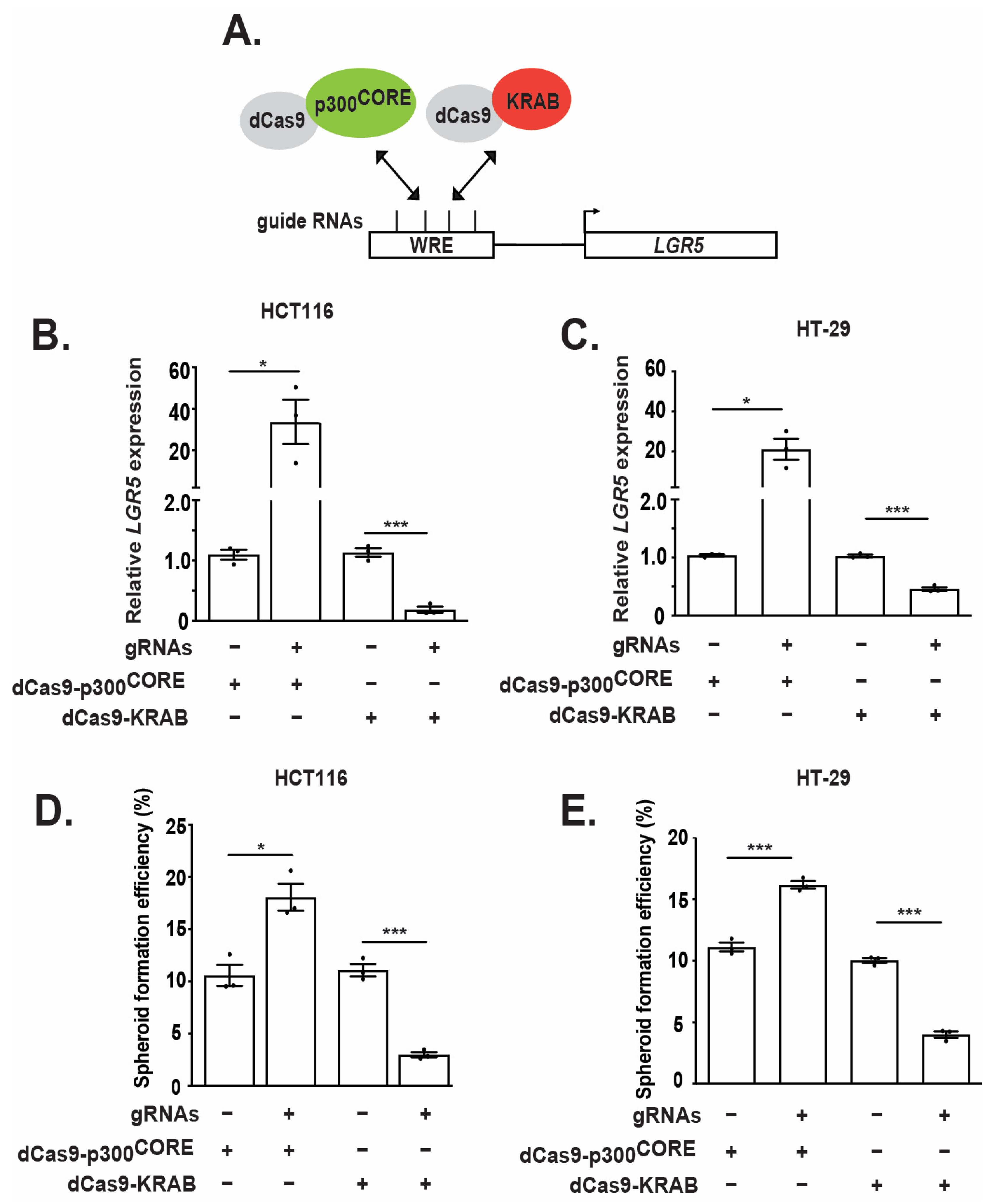
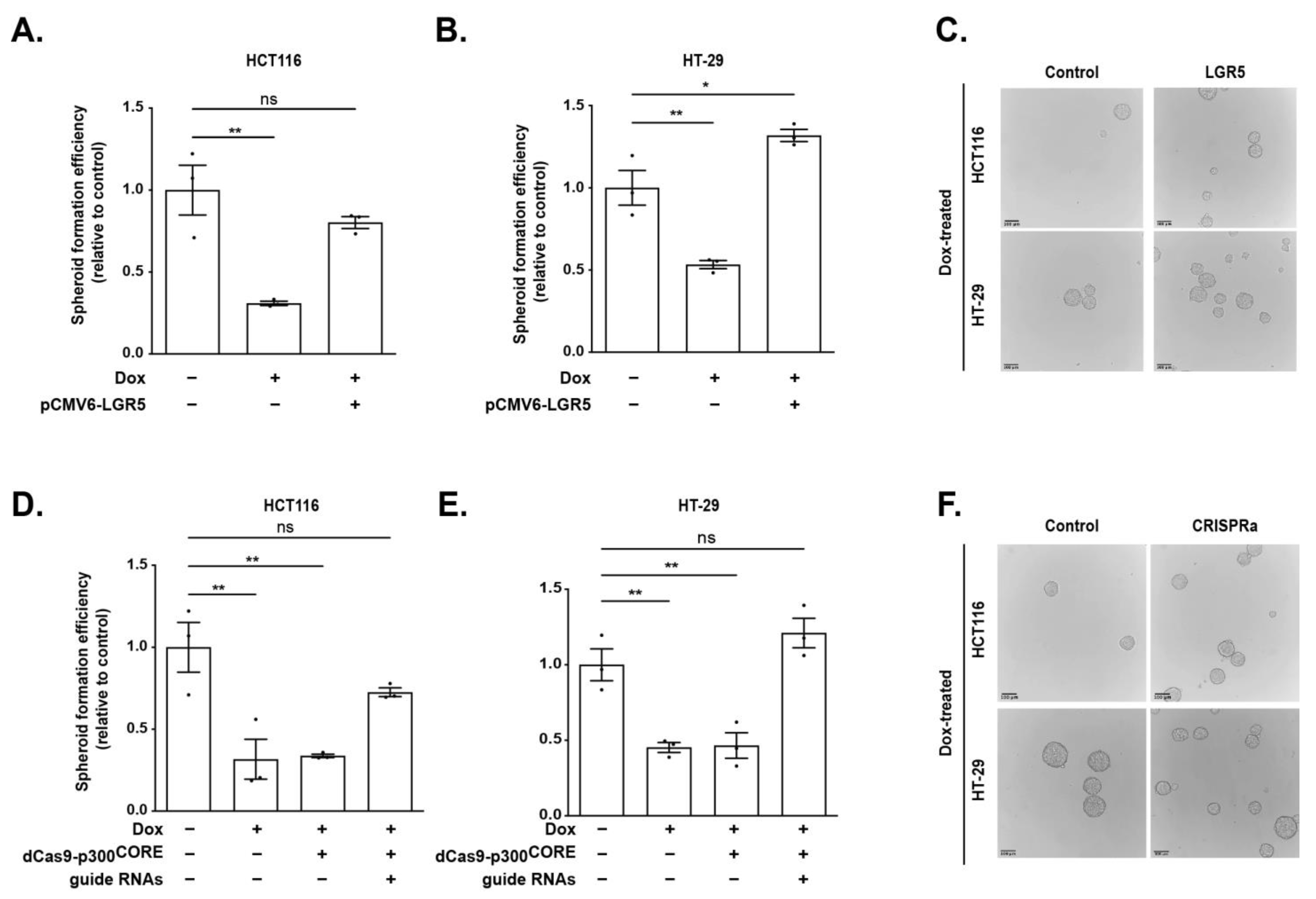
3.5. Epigenetic Regulation of the WRE Impacts LGR5 Expression and Spheroid Formation
3.6. LGR5 Rescues TCF7L1-Mediated Reduction in Spheroid Formation Efficiency
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Muzny, D.M.; Dinh, H.H.; Lewis, L.R.; Morgan, M.B.; Santibanez, J.; Shinbrot, E.; Trevino, L.R.; Wu, Y.-Q.; Wang, M.; Donehower, L.A.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Barker, N. Adult intestinal stem cells: Critical drivers of epithelial homeostasis and regeneration. Nat. Rev. Mol. Cell Biol. 2014, 15, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Gehart, H.; Clevers, H. Tales from the crypt: New insights into intestinal stem cells. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/β-Catenin Signaling and Disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Cadigan, K.M.; Waterman, M.L. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb. Perspect. Biol. 2012, 4, a007906. [Google Scholar] [CrossRef]
- Archbold, H.C.; Yang, Y.X.; Chen, L.; Cadigan, K.M. How do they do Wnt they do?: Regulation of transcription by the Wnt/β-catenin pathway. Acta Physiol. 2012, 204, 74–109. [Google Scholar] [CrossRef]
- Eshelman, M.A.; Shah, M.; Raup-Konsavage, W.M.; Rennoll, S.A.; Yochum, G.S. TCF7L1 recruits CtBP and HDAC1 to repress DICKKOPF4 gene expression in human colorectal cancer cells. Biochem. Biophys. Res. Commun. 2017, 487, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.; Chatterjee, S.S.; Jain, S.; Katari, M.; Dasgupta, R. TCF7L1 Modulates Colorectal Cancer Growth by Inhibiting Expression of the Tumor-Suppressor Gene EPHB3. Sci. Rep. 2016, 6, 28299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.; Rennoll, S.A.; Raup-Konsavage, W.M.; Yochum, G.S. A dynamic exchange of TCF3 and TCF4 transcription factors controls MYC expression in colorectal cancer cells. Cell Cycle 2015, 14, 323–332. [Google Scholar] [CrossRef]
- van de Wetering, M.; Sancho, E.; Verweij, C.; de Lau, W.; Oving, I.; Hurlstone, A.; van der Horn, K.; Batlle, E.; Coudreuse, D.; Haramis, A.-P.; et al. The β-Catenin/TCF-4 Complex Imposes a Crypt Progenitor Phenotype on Colorectal Cancer Cells. Cell 2002, 111, 241–250. [Google Scholar] [CrossRef]
- Barker, N.; Van Es, J.H.; Kuipers, J.; Kujala, P.; Van Den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, D.; Barker, N.; McNeil, N.; Hu, Y.; Camps, J.; McKinnon, K.; Clevers, H.; Ried, T.; Gaiser, T. LGR5 positivity defines stem-like cells in colorectal cancer. Carcinogenesis 2014, 35, 849–858. [Google Scholar] [CrossRef]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.L.; Begthel, H.L.; van den Born, M.M.W.; Danenberg, E.M.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef]
- Tetteh, P.W.; Basak, O.; Farin, H.F.; Wiebrands, K.; Kretzschmar, K.; Begthel, H.; Born, M.V.D.; Korving, J.; de Sauvage, F.; van Es, J.H.; et al. Replacement of Lost Lgr5-Positive Stem Cells through Plasticity of Their Enterocyte-Lineage Daughters. Cell Stem Cell 2016, 18, 203–213. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, E.; Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; et al. A distinct role for Lgr5 + stem cells in primary and metastatic colon cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Lin, W.; Wen, L.; Li, G. Lgr5 in cancer biology: Functional identification of Lgr5 in cancer progression and potential opportunities for novel therapy. Stem Cell Res. Ther. 2019, 10, 219. [Google Scholar] [CrossRef]
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.; Sugimoto, S.; Kanai, T.; Sato, T. Visualization and targeting of LGR5 + human colon cancer stem cells. Nature 2017, 545, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, A.; Oost, K.C.; Kester, L.; Morgner, J.; Bornes, L.; Bruens, L.; Spaargaren, L.; Azkanaz, M.; Schelfhorst, T.; Beerling, E.; et al. Plasticity of Lgr5-Negative Cancer Cells Drives Metastasis in Colorectal Cancer. Cell Stem Cell 2020, 26, 569–578.e567. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.-H.; Noah, T.K.; Chen, M.-S.; Zou, W.; Borras, E.; Vilar, E.; Shroyer, N.F. SPDEF Induces Quiescence of Colorectal Cancer Cells by Changing the Transcriptional Targets of β-catenin. Gastroenterology 2017, 153, 205–218.e208. [Google Scholar] [CrossRef] [PubMed]
- Grinat, J.; Heuberger, J.; Vidal, R.O.; Goveas, N.; Kosel, F.; Berenguer-Llergo, A.; Kranz, A.; Wulf-Goldenberg, A.; Behrens, D.; Melcher, B.; et al. The epigenetic regulator Mll1 is required for Wnt-driven intestinal tumorigenesis and cancer stemness. Nat. Commun. 2020, 11, 6422. [Google Scholar] [CrossRef]
- Matthews, S.M.; Eshelman, M.A.; Berg, A.S.; Koltun, W.A.; Yochum, G.S. The Crohn’s disease associated SNP rs6651252 impacts MYC gene expression in human colonic epithelial cells. PLoS ONE 2019, 14, e0212850. [Google Scholar] [CrossRef] [PubMed]
- Konsavage, W.M.; Kyler, S.L.; Rennoll, S.A.; Jin, G.; Yochum, G.S. Wnt/β-Catenin Signaling Regulates Yes-associated Protein (YAP) Gene Expression in Colorectal Carcinoma Cells. J. Biol. Chem. 2012, 287, 11730–11739. [Google Scholar] [CrossRef] [PubMed]
- Rennoll, S.A.; Eshelman, M.A.; Raup-Konsavage, W.M.; Kawasawa, Y.I.; Yochum, G.S. The MYC 3′ Wnt-responsive element drives oncogenic MYC expression in human colorectal cancer cells. Cancers 2016, 8, 52. [Google Scholar] [CrossRef] [PubMed]
- Yochum, G.S.; Cleland, R.; Goodman, R.H. A Genome-Wide Screen for β-Catenin Binding Sites Identifies a Downstream Enhancer Element That Controls c-Myc Gene Expression. Mol. Cell. Biol. 2008, 28, 7368–7379. [Google Scholar] [CrossRef]
- Concordet, J.-P.; Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Yochum, G.S.; McWeeney, S.; Rajaraman, V.; Cleland, R.; Peters, S.; Goodman, R.H. Serial analysis of chromatin occupancy identifies β-catenin target genes in colorectal carcinoma cells. Proc. Natl. Acad. Sci. USA 2007, 104, 3324–3329. [Google Scholar] [CrossRef] [PubMed]
- Bottomly, D.; Kyler, S.L.; McWeeney, S.K.; Yochum, G.S. Identification of β-catenin binding regions in colon cancer cells using ChIP-Seq. Nucleic Acids Res. 2010, 38, 5735–5745. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, S.; Ahmed, M.; Lorenzi, F.; Nateri, A.S. Spheroid-Formation (Colonosphere) Assay for in Vitro Assessment and Expansion of Stem Cells in Colon Cancer. Stem Cell Rev. Rep. 2016, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Kemper, K.; Prasetyanti, P.R.; De Lau, W.; Rodermond, H.; Clevers, H.; Medema, J.P. Monoclonal antibodies against Lgr5 identify human colorectal cancer stem cells. Stem Cells 2012, 30, 2378–2386. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Badran, A.H.; Liu, D.R. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 2017, 169, 559. [Google Scholar] [CrossRef] [PubMed]
- Hrckulak, D.; Kolar, M.; Strnad, H.; Korinek, V. TCF/LEF transcription factors: An update from the internet resources. Cancers 2016, 8, 70. [Google Scholar] [CrossRef]
- Mayer, C.-D.; de La Giclais, S.M.; Alsehly, F.; Hoppler, S. Diverse lef/tcf expression in human colorectal cancer correlates with altered wnt-regulated transcriptome in a meta-analysis of patient biopsies. Genes 2020, 11, 538. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; He, L.; Cao, B.; Zhao, X.; Zhang, K.; Li, Y.; Beck, P.; Zhou, Z.; Tian, Y.; Cheng, S.; et al. Differential expression of LEF1/TCFs family members in colonic carcinogenesis: LEF1/TCFs in colonic carcinogenesis. Mol. Carcinog. 2017, 56, 2372–2381. [Google Scholar] [CrossRef] [PubMed]
- Hua, F.; Shang, S.; Yang, Y.-W.; Zhang, H.-Z.; Xu, T.-L.; Yu, J.-J.; Zhou, D.-D.; Cui, B.; Li, K.; Lv, X.-X.; et al. TRIB3 Interacts with β-Catenin and TCF4 to Increase Stem Cell Features of Colorectal Cancer Stem Cells and Tumorigenesis. Gastroenterology 2019, 156, 708–721.e715. [Google Scholar] [CrossRef] [PubMed]
- Whissell, G.; Montagni, E.; Martinelli, P.; Hernando-Momblona, X.; Sevillano, M.; Jung, P.; Cortina, C.; Calon, A.; Abuli, A.; Castells, A.; et al. The transcription factor GATA6 enables self-renewal of colon adenoma stem cells by repressing BMP gene expression. Nat. Cell Biol. 2014, 16, 695–707. [Google Scholar] [CrossRef]
- Tsuji, S.; Kawasaki, Y.; Furukawa, S.; Taniue, K.; Hayashi, T.; Okuno, M.; Hiyoshi, M.; Kitayama, J.; Akiyama, T. The miR-363-GATA6-Lgr5 pathway is critical for colorectal tumourigenesis. Nat. Commun. 2014, 5, 3150. [Google Scholar] [CrossRef]
- Jian, Y.; Wang, M.; Zhang, Y.; Ou, R.; Zhu, Z.; Ou, Y.; Chen, X.; Liang, X.; Ding, Y.; Song, L.; et al. Jade family PHD finger 3 (JADE3) increases cancer stem cell-like properties and tumorigenicity in colon cancer. Cancer Lett. 2018, 428, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Ikeda, N.; Miyashita, K.; Nuriya, H.; Hara, T. DEAD box protein DDX1 promotes colorectal tumorigenesis through transcriptional activation of the LGR5 gene. Cancer Sci. 2018, 109, 2479–2489. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Jeong, J.; Choi, J.; Lim, J.; Dinesh, R.K.; Braverman, J.; Hong, J.Y.; Maher, S.E.; Vesely, M.C.A.; Kim, W.; et al. Dickkopf-2 regulates the stem cell marker LGR5 in colorectal cancer via HNF4 alpha 1. iScience 2021, 24, 102411. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, E.G.; Nasreddin, N.; Valbuena, G.N.; Mulholland, E.J.; Belnoue-Davis, H.L.; Eggington, H.R.; Schenck, R.O.; Wouters, V.M.; Wirapati, P.; Gilroy, K.; et al. Dynamic and adaptive cancer stem cell population admixture in colorectal neoplasia. Cell Stem Cell 2022, 29, 1213–1228.e1218. [Google Scholar] [CrossRef] [PubMed]
- Zeuner, A.; Todaro, M.; Stassi, G.; De Maria, R. Colorectal cancer stem cells: From the crypt to the clinic. Cell Stem Cell 2014, 15, 692–705. [Google Scholar] [CrossRef] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
King, C.M.; Marx, O.M.; Ding, W.; Koltun, W.A.; Yochum, G.S. TCF7L1 Regulates LGR5 Expression in Colorectal Cancer Cells. Genes 2023, 14, 481. https://doi.org/10.3390/genes14020481
King CM, Marx OM, Ding W, Koltun WA, Yochum GS. TCF7L1 Regulates LGR5 Expression in Colorectal Cancer Cells. Genes. 2023; 14(2):481. https://doi.org/10.3390/genes14020481
Chicago/Turabian StyleKing, Carli M., Olivia M. Marx, Wei Ding, Walter A. Koltun, and Gregory S. Yochum. 2023. "TCF7L1 Regulates LGR5 Expression in Colorectal Cancer Cells" Genes 14, no. 2: 481. https://doi.org/10.3390/genes14020481