
Genetic Factors Implicated in the Investigation of Possible Connections between Alzheimer’s Disease and Primary Open Angle Glaucoma

Abstract
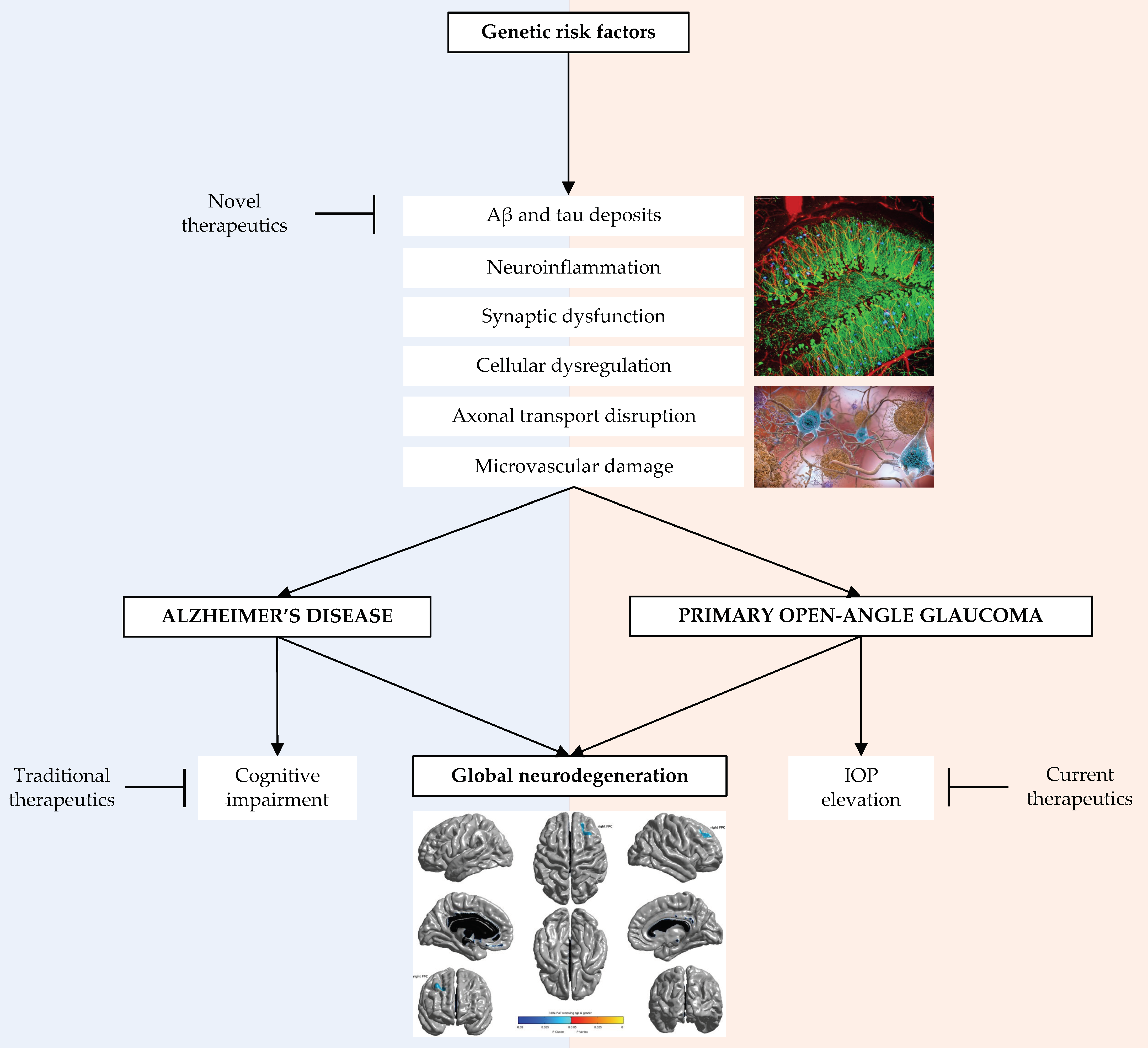
:1. Introduction
2. Overview of Alzheimer’s Disease and Glaucoma
2.1. Alzheimer’s Disease
2.2. Glaucoma
3. Background of Connections between Alzheimer’s Disease and Glaucoma
4. Genetic Factors Relevant to Alzheimer’s Disease and Primary Open Angle Glaucoma
5. Implications of Genetic Connections between Alzheimer’s Disease and Glaucoma
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mayeux, R.; Stern, Y. Epidemiology of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allison, K.; Patel, D.; Alabi, O. Epidemiology of Glaucoma: The Past, Present, and Predictions for the Future. Cureus 2020, 12, e11686. [Google Scholar] [CrossRef] [PubMed]
- Casson, R.J.; Chidlow, G.; Wood, J.P.M.; Crowston, J.G.; Goldberg, I. Definition of glaucoma: Clinical and experimental concepts. Clin. Exp. Ophthalmol. 2012, 40, 341–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, D.; Nordberg, A.; Westman, E. Biological subtypes of Alzheimer disease: A systematic review and meta-analysis. Neurology 2020, 94, 436–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennon, J.C.; Aita, S.L.; Del Bene, V.A.; Rhoads, T.; Resch, Z.J.; Eloi, J.M.; Walker, K.A. Black and White individuals differ in dementia prevalence, risk factors, and symptomatic presentation. Alzheimer’s Dement. 2021, 18, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Rudnicka, A.R.; Mt-Isa, S.; Owen, C.; Cook, D.; Ashby, D. Variations in Primary Open-Angle Glaucoma Prevalence by Age, Gender, and Race: A Bayesian Meta-Analysis. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4254–4261. [Google Scholar] [CrossRef] [Green Version]
- Allen, K.F.; Gaier, E.D.; Wiggs, J.L. Genetics of Primary Inherited Disorders of the Optic Nerve: Clinical Applications. Cold Spring Harb. Perspect. Med. 2015, 5, a017277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.W.; Chan, N.C.; Sadun, A.A. Glaucoma as Neurodegeneration in the Brain. Eye Brain 2021, 13, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Cho, K.-S.; Thee, E.F.; Jager, M.J.; Chen, D.F. Neuroinflammation and microglia in glaucoma: Time for a paradigm shift. J. Neurosci. Res. 2018, 97, 70–76. [Google Scholar] [CrossRef] [Green Version]
- Mroczkowska, S.; Shokr, H.; Benavente-Pérez, A.; Negi, A.; Bentham, P.; Gherghel, D. Retinal Microvascular Dysfunction Occurs Early and Similarly in Mild Alzheimer’s Disease and Primary-Open Angle Glaucoma Patients. J. Clin. Med. 2022, 11, 6702. [Google Scholar] [CrossRef]
- Jones-Odeh, E.; Hammond, C.J. How strong is the relationship between glaucoma, the retinal nerve fibre layer, and neurodegenerative diseases such as Alzheimer’s disease and multiple sclerosis? Eye 2015, 29, 1270–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernowo, A.T.; Boucard, C.C.; Jansonius, N.M.; Hooymans, J.M.M.; Cornelissen, F.W. Automated Morphometry of the Visual Pathway in Primary Open-Angle Glaucoma. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2758–2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellenguez, C.; Küçükali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef] [PubMed]
- Schuster, A.K.; Erb, C.; Hoffmann, E.M.; Dietlein, T.; Pfeiffer, N. The Diagnosis and Treatment of Glaucoma. Dtsch. Ärzteblatt Int. 2020, 117, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Doucette, L.P.; Rasnitsyn, A.; Seifi, M.; Walter, M.A. The interactions of genes, age, and environment in glaucoma pathogenesis. Surv. Ophthalmol. 2015, 60, 310–326. [Google Scholar] [CrossRef] [PubMed]
- Vaz, M.; Silvestre, S. Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharmacol. 2020, 887, 173554. [Google Scholar] [CrossRef] [PubMed]
- Steinman, J.; Sun, H.-S.; Feng, Z.-P. Microvascular Alterations in Alzheimer’s Disease. Front. Cell. Neurosci. 2021, 14, 618986. [Google Scholar] [CrossRef]
- Gogineni, A. Brain Showing Hallmarks of Alzheimer’s Disease (Plaques in Blue). Flickr. Available online: https://www.flickr.com/photos/nihgov/26709211161/ (accessed on 18 January 2023).
- National Institute on Aging, NIH. β-Amyloid Plaques and Tau in the Brain. Flickr. Available online: https://www.flickr.com/photos/nihgov/38686503251/ (accessed on 18 January 2023).
- Wang, J.; Li, T.; Sabel, B.A.; Chen, Z.; Wen, H.; Li, J.; Xie, X.; Yang, D.; Chen, W.; Wang, N.; et al. Structural brain alterations in primary open angle glaucoma: A 3T MRI study. Sci. Rep. 2016, 6, 18969. [Google Scholar] [CrossRef]
- Gharahkhani, P.; Jorgenson, E.; Hysi, P.; Khawaja, A.P.; Pendergrass, S.; Han, X.; Ong, J.S.; Hewitt, A.W.; Segrè, A.V.; Rouhana, J.M.; et al. Genome-wide meta-analysis identifies 127 open-angle glaucoma loci with consistent effect across ancestries. Nat. Commun. 2021, 12, 1258. [Google Scholar] [CrossRef]
- Zheng, C.; Liu, S.; Zhang, X.; Hu, Y.; Shang, X.; Zhu, Z.; Huang, Y.; Wu, G.; Xiao, Y.; Du, Z.; et al. Shared genetic architecture between the two neurodegenerative diseases: Alzheimer’s disease and glaucoma. Front. Aging Neurosci. 2022, 14, 880576. [Google Scholar] [CrossRef]
- Volicer, L.; McKee, A.; Hewitt, S. Dementia. Neurol. Clin. 2001, 19, 867–885. [Google Scholar] [CrossRef] [PubMed]
- Gale, S.A.; Acar, D.; Daffner, K.R. Dementia. Am. J. Med. 2018, 131, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O. Biomarkers for neurodegenerative diseases. Nat. Med. 2021, 27, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.F. Early-Onset Alzheimer Disease. Neurol. Clin. 2017, 35, 263–281. [Google Scholar] [CrossRef] [Green Version]
- De Ture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32–35. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s Disease: Pathogenesis, Diagnostics, and Therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [Green Version]
- Weller, J.; Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 2018, 7, 1161. [Google Scholar] [CrossRef] [Green Version]
- Davis, B.M.; Crawley, L.; Pahlitzsch, M.; Javaid, F.; Cordeiro, M.F. Glaucoma: The retina and beyond. Acta Neuropathol. 2016, 132, 807–826. [Google Scholar] [CrossRef] [Green Version]
- Havvas, I.; Papaconstantinou, D.; Moschos, M.M.; Theodossiadis, P.G.; Andreanos, V.; Ekatomatis, P.; Vergados, I.; Andreanos, D. Comparison of SWAP and SAP on the point of glaucoma conversion. Clin. Ophthalmol. 2013, 7, 1805–1810. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Jeong, H.J.; Lee, J.H.; Kim, Y.J.; Kim, E.Y.; Kim, Y.Y.; Ryu, T.; Cho, Z.-H.; Kim, Y.-B. An Investigation of Lateral Geniculate Nucleus Volume in Patients With Primary Open-Angle Glaucoma Using 7 Tesla Magnetic Resonance Imaging. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3468–3476. [Google Scholar] [CrossRef]
- Baiocchi, S.; Mazzotta, C.; Sgheri, A.; Di Maggio, A.; Bagaglia, S.A.; Posarelli, M.; Ciompi, L.; Meduri, A.; Tosi, G.M. In vivo confocal microscopy: Qualitative investigation of the conjunctival and corneal surface in open angle glaucomatous patients undergoing the XEN-Gel implant, trabeculectomy or medical therapy. Eye Vis. 2020, 7, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FFrisina, R.; Meduri, A. Intraoperative real-time image-guided ab externo canaloplasty. Eye 2019, 33, 1510–1513. [Google Scholar] [CrossRef] [PubMed]
- Bales, T.R.; Lopez, M.J.; Clark, J. Embryology, Eye. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK538480/ (accessed on 26 November 2022).
- Zabel, P.; Kałużny, J.J.; Wiłkość-Dębczyńska, M.; Gębska-Tołoczko, M.; Suwała, K.; Kucharski, R.; Araszkiewicz, A. Peripapillary Retinal Nerve Fiber Layer Thickness in Patients with Alzheimer’s Disease: A Comparison of Eyes of Patients with Alzheimer’s Disease, Primary Open-Angle Glaucoma, and Preperimetric Glaucoma and Healthy Controls. Med. Sci. Monit. 2019, 25, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.S.; Ritch, R.; Schwartz, B.; Lee, S.S.; Miller, N.R.; Chi, T.; Hsieh, F.Y. Optic Nerve Head and Nerve Fiber Layer in Alzheimer’s Disease. Arch. Ophthalmol. 1991, 109, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Bevan, R.J.; Hughes, T.R.; Williams, P.A.; Good, M.A.; Morgan, B.P.; Morgan, J.E. Retinal ganglion cell degeneration correlates with hippocampal spine loss in experimental Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 216. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.-M.; Zhou, Q.; Liu, X.-Y.; Shi, C.-Z.; Chen, J.; Huang, X.-H. Structural and functional brain changes in early- and mid-stage primary open-angle glaucoma using voxel-based morphometry and functional magnetic resonance imaging. Medicine 2017, 96, e6139. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Liao, H.; Chen, H.; Deng, S.; Jianxian, L.; Deng, C.; Lin, J.; Ge, J.; Zhuo, Y. Elevated Intraocular Pressure Induces Amyloid-β Deposition and Tauopathy in the Lateral Geniculate Nucleus in a Monkey Model of Glaucoma. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5434–5443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinnon, S.J.; Lehman, D.M.; A Kerrigan-Baumrind, L.; A Merges, C.; Pease, M.; Kerrigan, D.F.; Ransom, N.L.; Tahzib, N.G.; Reitsamer, H.A.; Levkovitch-Verbin, H.; et al. Caspase activation and amyloid precursor protein cleavage in rat ocular hypertension. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1077–1087. [Google Scholar]
- Sen, S.; Saxena, R.; Tripathi, M.; Vibha, D.; Dhiman, R. Neurodegeneration in Alzheimer’s disease and glaucoma: Overlaps and missing links. Eye 2020, 34, 1546–1553. [Google Scholar] [CrossRef]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired Insulin and Insulin-Like Growth Factor Expression and Signaling Mechanisms in Alzheimer’s Disease—Is this Type 3 Diabetes? J. Alzheimer’s Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.W.; Lee, S.; Park, C.; Kim, D.J. Elevated intraocular pressure is associated with insulin resistance and metabolic syndrome. Diabetes/Metabolism Res. Rev. 2005, 21, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Hernández, J.C.C.; Bracko, O.; Kersbergen, C.J.; Muse, V.; Haft-Javaherian, M.; Berg, M.; Park, L.; Vinarcsik, L.K.; Ivasyk, I.; Rivera, D.A.; et al. Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nat. Neurosci. 2019, 22, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukhari, S.M.I.; Yew, K.K.; Thambiraja, R.; Sulong, S.; Rasool, A.H.G.; Tajudin, L.-S.A. Microvascular endothelial function and primary open angle glaucoma. Ther. Adv. Ophthalmol. 2019, 11, 2515841419868100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabel, P.; Kaluzny, J.J.; Wilkosc-Debczynska, M.; Gebska-Toloczko, M.; Suwala, K.; Zabel, K.; Zaron, A.; Kucharski, R.; Araszkiewicz, A. Comparison of Retinal Microvasculature in Patients With Alzheimer’s Disease and Primary Open-Angle Glaucoma by Optical Coherence Tomography Angiography. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3447–3455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullany, S.; Xiao, L.; Qassim, A.; Marshall, H.; Gharahkhani, P.; MacGregor, S.; Hassall, M.M.; Siggs, O.M.; Souzeau, E.; Craig, J.E. Normal-tension glaucoma is associated with cognitive impairment. Br. J. Ophthalmol. 2021, 106, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-W.; Lin, C.-C.; Kao, C.-H.; Chen, H.-Y. Association Between Glaucoma and the Risk of Dementia. Medicine 2016, 95, e2833. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zou, J.; Geng, W.; Wang, A. Association between glaucoma and the risk of Alzheimer’s disease: A systematic review of observational studies. Acta Ophthalmol. 2019, 97, 665–671. [Google Scholar] [CrossRef]
- Lin, I.-C.; Wang, Y.-H.; Wang, T.-J.; Wang, I.-J.; Shen, Y.-D.; Chi, N.-F.; Chien, L.-N. Glaucoma, Alzheimer’s Disease, and Parkinson’s Disease: An 8-Year Population-Based Follow-Up Study. PLoS ONE 2014, 9, e108938. [Google Scholar] [CrossRef]
- Kessing, L.V.; Lopez, A.G.; Andersen, P.K.; Kessing, S.V. No Increased Risk of Developing Alzheimer Disease in Patients With Glaucoma. Eur. J. Gastroenterol. Hepatol. 2007, 16, 47–51. [Google Scholar] [CrossRef]
- Zhao, W.; Lv, X.; Wu, G.; Zhou, X.; Tian, H.; Qu, X.; Sun, H.; He, Y.; Zhang, Y.; Wang, C.; et al. Glaucoma Is Not Associated With Alzheimer’s Disease or Dementia: A Meta-Analysis of Cohort Studies. Front. Med. 2021, 8, 688551. [Google Scholar] [CrossRef]
- Belamkar, A.V.; Mansukhani, S.A.; Savica, R.; Spiegel, M.R.; Hodge, D.O.; Sit, A.J. Incidence of Dementia in Patients With Open-angle Glaucoma: A Population-based Study. Eur. J. Gastroenterol. Hepatol. 2020, 30, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Lang, L.; Clifford, A.; Wei, L.; Zhang, D.; Leung, D.; Augustine, G.; Danat, I.M.; Zhou, W.; Copeland, J.R.; Anstey, K.J.; et al. Prevalence and determinants of undetected dementia in the community: A systematic literature review and a meta-analysis. BMJ Open 2017, 7, e011146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leite, M.T.; Sakata, L.M.; Medeiros, F.A. Managing glaucoma in developing countries. Arq. Bras. Oftalmol. 2011, 74, 83–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topouzis, F.; Coleman, A.L.; Harris, A.; Koskosas, A.; Founti, P.; Gong, G.; Yu, F.; Anastasopoulos, E.; Pappas, T.; Wilson, M.R. Factors Associated with Undiagnosed Open-Angle Glaucoma: The Thessaloniki Eye Study. Am. J. Ophthalmol. 2008, 145, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Bradford, A.; Kunik, M.E.; Schulz, P.; Williams, S.P.; Singh, H. Missed and Delayed Diagnosis of Dementia in Primary Care: Prevalence and Contributing Factors. Alzheimer Dis. Assoc. Disord. 2009, 23, 306–314. [Google Scholar] [CrossRef]
- Hysi, P.G.; Cheng, C.-Y.; Springelkamp, H.; Macgregor, S.; Bailey, J.N.C.; Wojciechowski, R.; Vitart, V.; Nag, A.; Hewitt, A.W.; Höhn, R.; et al. Genome-wide analysis of multi-ancestry cohorts identifies new loci influencing intraocular pressure and susceptibility to glaucoma. Nat. Genet. 2014, 46, 1126–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, J.-M.; Spickett, C.M. The role of CELF proteins in neurological disorders. RNA Biol. 2010, 7, 474–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rustenhoven, J.; Smith, A.M.; Smyth, L.C.; Jansson, D.; Scotter, E.L.; Swanson, M.E.V.; Aalderink, M.; Coppieters, N.; Narayan, P.; Handley, R.; et al. PU.1 regulates Alzheimer’s disease-associated genes in primary human microglia. Mol. Neurodegener. 2018, 13, 44. [Google Scholar] [CrossRef] [Green Version]
- Celada, A.; Borras, F.E.; Soler, C.; Lloberas, J.; Klemsz, M.; van Beveren, C.; McKercher, S.; Maki, R.A. The transcription factor PU.1 is involved in macrophage proliferation. J. Exp. Med. 1996, 184, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.E.; Andrews, R.; Holmans, P.; Hill, M.; Taylor, P.R. Modest changes in Spi1 dosage reveal the potential for altered microglial function as seen in Alzheimer’s disease. Sci. Rep. 2021, 11, 14935. [Google Scholar] [CrossRef] [PubMed]
- Chiasseu, M.; Vargas, J.L.C.; Destroismaisons, L.; Velde, C.V.; Leclerc, N.; Di Polo, A. Tau Accumulation, Altered Phosphorylation, and Missorting Promote Neurodegeneration in Glaucoma. J. Neurosci. 2016, 36, 5785–5798. [Google Scholar] [CrossRef] [Green Version]
- Chi, H.; Yao, R.; Sun, C.; Leng, B.; Shen, T.; Wang, T.; Zhang, S.; Li, M.; Yang, Y.; Sun, H.; et al. Blood Neuroexosomal Mitochondrial Proteins Predict Alzheimer Disease in Diabetes. Diabetes 2022, 71, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Strang, K.H.; Golde, T.E.; Giasson, B.I. MAPT Mutations, Tauopathy, and Mechanisms of Neurodegeneration. Lab. Investig. 2019, 99, 912–928. [Google Scholar] [CrossRef] [PubMed]
- Kröll-Hermi, A.; Ebstein, F.; Stoetzel, C.; Geoffroy, V.; Schaefer, E.; Scheidecker, S.; Bär, S.; Takamiya, M.; Kawakami, K.; Zieba, B.A.; et al. Proteasome subunit PSMC3 variants cause neurosensory syndrome combining deafness and cataract due to proteotoxic stress. EMBO Mol. Med. 2020, 12, e11861. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Liu, X.; Yu, W.-M.; Liu, J.; Nibbelink, M.G.; Guo, C.; Finkel, T.; Qu, C.-K. A Critical Role of Mitochondrial Phosphatase Ptpmt1 in Embryogenesis Reveals a Mitochondrial Metabolic Stress-Induced Differentiation Checkpoint in Embryonic Stem Cells. Mol. Cell. Biol. 2011, 31, 4902–4916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banwell, B.L.; Ohno, K.; Sieb, J.P.; Engel, A.G. Novel truncating RAPSN mutations causing congenital myasthenic syndrome responsive to 3,4-diaminopyridine. Neuromuscul. Disord. 2004, 14, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Wang, J.; Liu, C.; Jiang, T.; Yang, N.; Liu, D.; Zhao, H.; Xu, Z. Zinc transporter SLC39A13/ZIP13 facilitates the metastasis of human ovarian cancer cells via activating Src/FAK signaling pathway. J. Exp. Clin. Cancer Res. 2021, 40, 199. [Google Scholar] [CrossRef]
- Preman, P.; Alfonso-Triguero, M.; Alberdi, E.; Verkhratsky, A.; Arranz, A. Astrocytes in Alzheimer’s Disease: Pathological Significance and Molecular Pathways. Cells 2021, 10, 540. [Google Scholar] [CrossRef] [PubMed]
- Choquet, H.; Paylakhi, S.; Kneeland, S.C.; Thai, K.K.; Hoffmann, T.J.; Yin, J.; Kvale, M.N.; Banda, Y.; Tolman, N.G.; Williams, P.A.; et al. A multiethnic genome-wide association study of primary open-angle glaucoma identifies novel risk loci. Nat. Commun. 2018, 9, 2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zagajewska, K.; Piątkowska, M.; Goryca, K.; Bałabas, A.; Kluska, A.; Paziewska, A.; Pośpiech, E.; Grabska-Liberek, I.; Hennig, E.E. GWAS links variants in neuronal development and actin remodeling related loci with pseudoexfoliation syndrome without glaucoma. Exp. Eye Res. 2017, 168, 138–148. [Google Scholar] [CrossRef]
- Sharoukhov, D.; Bucinca-Cupallari, F.; Lim, H. Microtubule Imaging Reveals Cytoskeletal Deficit Predisposing the Retinal Ganglion Cell Axons to Atrophy in DBA/2J. Investig. Ophthalmol. Vis. Sci. 2018, 59, 5292–5300. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, A.; Aloni, E.; Korkotian, E.; Zaltsman, Y.; Oni-Biton, E.; Kuperman, Y.; Tsoory, M.; Shachnai, L.; Levin-Zaidman, S.; Brenner, O.; et al. Loss of forebrain MTCH2 decreases mitochondria motility and calcium handling and impairs hippocampal-dependent cognitive functions. Sci. Rep. 2017, 7, srep44401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berezniuk, I.; Lyons, P.J.; Sironi, J.J.; Xiao, H.; Setou, M.; Angeletti, R.H.; Ikegami, K.; Fricker, L.D. Cytosolic carboxypeptidase 5 removes α- and γ-linked glutamates from tubulin. J. Biol. Chem. 2013, 288, 30445–30453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokin, G.B.; Lillo, C.; Falzone, T.L.; Brusch, R.G.; Rockenstein, E.; Mount, S.L.; Raman, R.; Davies, P.; Masliah, E.; Williams, D.S.; et al. Axonopathy and Transport Deficits Early in the Pathogenesis of Alzheimer’s Disease. Science 2005, 307, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhou, J.; Starr, C.; Mohns, E.J.; Li, Y.; Chen, E.P.; Yoon, Y.; Kellner, C.P.; Tanaka, K.; Wang, H.; et al. Preservation of vision after CaMKII-mediated protection of retinal ganglion cells. Cell 2021, 184, 4299–4314.e12. [Google Scholar] [CrossRef] [PubMed]
- Katsumata, Y.; Nelson, P.T.; Estus, S.; Fardo, D.W. Translating Alzheimer’s disease–associated polymorphisms into functional candidates: A survey of IGAP genes and SNPs. Neurobiol. Aging 2018, 74, 135–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.-T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.-L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154.e7, Correction in Neuron 2018, 98, 1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood–brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Margeta, M.A.; Yin, Z.; Madore, C.; Pitts, K.M.; Letcher, S.M.; Tang, J.; Jiang, S.; Gauthier, C.D.; Silveira, S.R.; Schroeder, C.M.; et al. Apolipoprotein E4 impairs the response of neurodegenerative retinal microglia and prevents neuronal loss in glaucoma. Immunity 2022, 55, 1627–1644.e7. [Google Scholar] [CrossRef]
- Margeta, M.A.; Letcher, S.M.; Igo, R.P.; Bailey, J.N.C.; Pasquale, L.R.; Haines, J.L.; Butovsky, O.; Wiggs, J.L.; for the NEIGHBORHOOD consortium. Association of APOE With Primary Open-Angle Glaucoma Suggests a Protective Effect for APOE ε4. Investig. Ophthalmol. Vis. Sci. 2020, 61, 3. [Google Scholar] [CrossRef] [PubMed]
- Beason-Held, L.L.; Goh, J.O.; An, Y.; Kraut, M.A.; O’Brien, R.J.; Ferrucci, L.; Resnick, S.M. Changes in Brain Function Occur Years before the Onset of Cognitive Impairment. J. Neurosci. 2013, 33, 18008–18014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, R.; Ye, M.; Xu, X. An updated meta-analysis: Apolipoprotein E genotypes and risk of primary open-angle glaucoma. Mol. Vis. 2014, 20, 1025–1036. [Google Scholar] [PubMed]
- Kumar, A.; Sidhu, J.; Goyal, A.; Tsao, J.W. Alzheimer Disease. In StatPearls; Updated 2022 Jun 5; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK499922/ (accessed on 26 November 2022).
- Nilsson, P.; Iwata, N.; Muramatsu, S.-I.; Tjernberg, L.O.; Winblad, B.; Saido, T.C. Gene therapy in Alzheimer’s disease—Potential for disease modification. J. Cell. Mol. Med. 2010, 14, 741–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiggs, J.L.; Pasquale, L.R. Genetics of glaucoma. Hum. Mol. Genet. 2017, 26, R21–R27. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gene | Name | Location | Protein Product | Function | Pathogenic Mutation | Association with AD | Association with POAG |
|---|---|---|---|---|---|---|---|
| AGBL2 | ATP/GTP-binding protein-like 2 | 11p11.2 | Cytosolic carboxypeptidase 2 (CCP2) enzyme | Catalyzes post-translational modification of ɑ-tubulin subunit | rs11604825 rs11602395 | Microtubular axonal transport alteration | IOP elevation |
| MAPT | Microtubule associated protein tau | 17q21.31 | Microtubule-associated protein tau (MAPT) | Encodes tau proteins and other mRNA products | Post-translational alternative splicing and modification | Tau structure isoform variations | Misbalance in glaucomatous RGC axons |
| SPI1 | Spi-1 proto-oncogene | 11p11.2 | PU.1 erythroblast transformation specific (ETS) transcription factor | Promotes gene expression of hematopoietic cell lineages | 28 SNP’s | Microglia activation | Microglia activation, IOP elevation |
| CELF1 | CUG triplet repeat, RNA binding protein 1 (CUGBP) embryonic lethal abnormal vision (Elav)-like family member 1 | 11p11.2 | CELF/BRUNOL protein | Regulates pre-mRNA splicing and mRNA expression | rs4752845 rs34958982 rs66749409 rs12798346 rs56400411 | Alternative splicing of tau protein | IOP elevation |
| FAM180B | Family with sequence similarity 180 member B | 11p11.2 | Family with sequence similarity 180 member B | Presently unknown function | rs11605348 | Expressed at higher levels in brain | IOP elevation |
| MTCH2 | Mitochondrial carrier 2 | 11p11.2 | Solute carrierfamily 25 (SLC25) family nuclear-encoded mitochondrial transporters | Involved in apoptotic pathway via recruitment of Bcl-2 family BID protein | rs4752856 rs4752856 rs4752856 | Disrupts mitochondrial motility, metabolism, and function | IOP elevation |
| MYBPC3 | Myosin binding protein C3 | 11p11.2 | Cardiac isoform of myosin binding protein C (MyBP-C) | Regulates cardiac striated muscle contraction | rs2856661 | May affect axonal growth and synaptic development | IOP elevation |
| NDUFS3 | NADH:ubiquinone oxidoreductase core subunit S3 | 11p11.2 | Iron-sulfur protein (IP) component in mitochondrial NADH:ubiquinone oxidoreductase (complex I) | Involved in mitochondrial electron transport chain and cellular functions | rs2030166 rs2030166 | Increased levels in early stages of mild cognitive impairment | IOP elevation |
| PSMC3 | Proteasome 26S subunit, ATPase 3 | 11p11.2 | 26S proteasome | Involved in ubiquitin-proteasome degradation system | rs11600581 | Increased cellular proteotoxic stress | IOP elevation |
| PTPMT1 | Protein tyrosine phosphatase mitochondrial 1 | 11p11.2 | Phosphatidylglycerophosphatase and protein-tyrosine phosphatase 1 | Involved in mitochondrial metabolic pathways and embryogenesis | rs56400411 rs7945473 | Mitochondrial dysfunction | IOP elevation |
| RAPSN | Receptor associated protein of the synapse | 11p11.2 | Receptor-associated protein of the synapse | Anchors postsynaptic nicotinic acetylcholine receptors | rs35705029 | Acetylcholine neurotransmitter signal alteration | IOP elevation |
| SLC39A13 | Solute carrier family 39 member 13 | 11p11.2 | Zinc transporter transmembrane protein | Facilitates zinc transport across membranes | rs755554 | Disturbance of zinc homeostasis | IOP elevation |
| CADM2 | Cell adhesion molecule 2 | 3p12.1 | Synaptic cell adhesion molecule 1 (SynCAM) | Interacts with cytoskeletal proteins for cellular support and structure | r71316816 rs13101042 rs2220243 | Impaired synaptic adhesion and maintenance | Expressed almost exclusively in brain and retina and found to have altered expression throughout all stages of glaucoma |
| APP | Amyloid beta precursor protein | 21q21.3 | Amyloid precursor protein (APP) | Acts as a transmembrane cell surface receptor and regulates neuronal functions | rs59892895 | Abnormal cleavage forms β-amyloid plaques | Increased abnormal expression in glaucoma models |
| APOE | Apolipoprotein E | 19q13.32 | Apolipoprotein E | Maintains lipid metabolism and balance in circulatory and peripheral systems | ε4 allele | Increased risk for disease, increased amount of β-amyloid plaques | Reduced risk for disease, decrease activation of microglia |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuang, G.; Salowe, R.; O’Brien, J. Genetic Factors Implicated in the Investigation of Possible Connections between Alzheimer’s Disease and Primary Open Angle Glaucoma. Genes 2023, 14, 338. https://doi.org/10.3390/genes14020338
Kuang G, Salowe R, O’Brien J. Genetic Factors Implicated in the Investigation of Possible Connections between Alzheimer’s Disease and Primary Open Angle Glaucoma. Genes. 2023; 14(2):338. https://doi.org/10.3390/genes14020338
Chicago/Turabian StyleKuang, Grace, Rebecca Salowe, and Joan O’Brien. 2023. "Genetic Factors Implicated in the Investigation of Possible Connections between Alzheimer’s Disease and Primary Open Angle Glaucoma" Genes 14, no. 2: 338. https://doi.org/10.3390/genes14020338