
Role of RPA Phosphorylation in the ATR-Dependent G2 Cell Cycle Checkpoint

Abstract
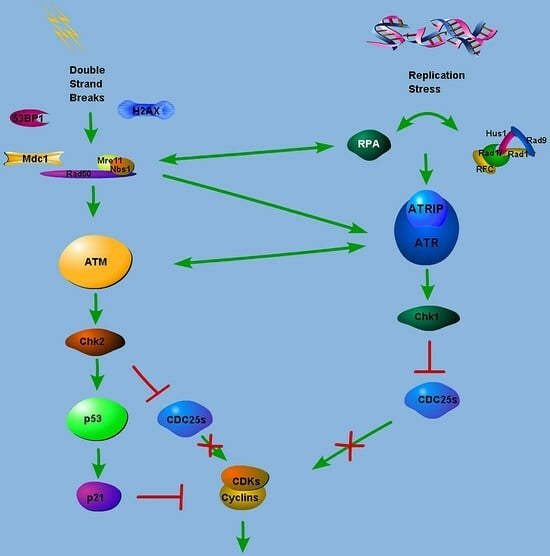
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. Synchronization of Cells and Treatment with Etoposide
2.3. Cell Lysates and Chromatin Isolation
2.4. Cloning and Creation of Transgenic Cell Lines
2.5. Immunoprecipitation and Immunoblotting
2.6. Flow Cytometry
2.7. Immunofluorescence Microscopy
3. Results
3.1. RPA Phosphorylation Affects ATR Signaling in G2 Synchronized Cells
3.2. Initial Chk1 Phosphorylation during DNA Damage in G2 Occurs Independently of RPA Phosphorylation
3.3. RPA Phosphorylation Promotes Chromatin Retention and Accumulation of TopBP1 and Rad9 in G2
3.4. RPA32 Ser33 Phosphorylation Indirectly Requires ATM Activity
3.5. RPA32 Ser4/Ser8 Phosphorylation Is Required for KAP-1 Phosphorylation
3.6. RPA32 Phosphorylation Influences Phosphorylation of H2AX and Rad51 Chromatin Loading
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Iliakis, G.; Wang, Y.; Guan, J.; Wang, H. DNA damage checkpoint control in cells exposed to ionizing radiation. Oncogene 2003, 22, 5834–5847. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Conrad, S.; Birraux, J.; Geuting, V.; Barton, O.; Ismail, A.; Kakarougkas, A.; Meek, K.; Taucher-Scholz, G.; Lobrich, M.; et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011, 30, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P. DNA resection in eukaryotes: Deciding how to fix the break. Nat. Struct. Mol. Biol. 2010, 17, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S.; Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef]
- Shiotani, B.; Zou, L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol. Cell 2009, 33, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef]
- Lee, J.; Kumagai, A.; Dunphy, W.G. The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J. Biol. Chem. 2007, 282, 28036–28044. [Google Scholar] [CrossRef]
- Lee, J.; Dunphy, W.G. Rad17 plays a central role in establishment of the interaction between TopBP1 and the Rad9-Hus1-Rad1 complex at stalled replication forks. Mol. Biol. Cell. 2010, 21, 926–935. [Google Scholar] [CrossRef]
- Chen, R.; Wold, M.S. Replication protein A: Single-stranded DNA’s first responder: Dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays 2014, 36, 1156–1161. [Google Scholar] [CrossRef]
- Oakley, G.G.; Patrick, S.M. Replication protein A: Directing traffic at the intersection of replication and repair. Front. Biosci. 2010, 15, 883–900. [Google Scholar] [CrossRef] [PubMed]
- Zernik-Kobak, M.; Vasunia, K.; Connelly, M.; Anderson, C.W.; Dixon, K. Sites of UV-induced phosphorylation of the p34 subunit of replication protein A from HeLa cells. J. Biol. Chem. 1997, 272, 23896–23904. [Google Scholar] [CrossRef] [PubMed]
- Niu, H.; Erdjument-Bromage, H.; Pan, Z.Q.; Lee, S.H.; Tempst, P.; Hurwitz, J. Mapping of amino acid residues in the p34 subunit of human single- stranded DNA-binding protein phosphorylated by DNA- dependent protein kinase and Cdc2 kinase in vitro. J. Biol. Chem. 1997, 272, 12634–12641. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Opiyo, S.O.; Manthey, K.; Glanzer, J.G.; Ashley, A.K.; Amerin, C.; Troksa, K.; Shrivastav, M.; Nickoloff, J.A.; Oakley, G.G. Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 2012, 40, 10780–10794. [Google Scholar] [CrossRef]
- Vassin, V.M.; Wold, M.S.; Borowiec, J.A. Replication Protein A (RPA) Phosphorylation Prevents RPA Association with Replication Centers. Mol. Cell. Biol. 2004, 24, 1930–1943. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 2015, 25, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Anantha, R.W.; Vassin, V.M.; Borowiec, J.A. Sequential and synergistic modification of human RPA stimulates chromosomal DNA repair. J. Biol. Chem. 2007, 282, 35910–35923. [Google Scholar] [CrossRef]
- Vassin, V.M.; Anantha, R.W.; Sokolova, E.; Kanner, S.; Borowiec, J.A. Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA-replication stress. J. Cell Sci. 2009, 122, 4070–4080. [Google Scholar] [CrossRef]
- Shiotani, B.; Nguyen, H.D.; Hakansson, P.; Marechal, A.; Tse, A.; Tahara, H.; Zou, L. Two Distinct Modes of ATR Activation Orchestrated by Rad17 and Nbs1. Cell Rep. 2013, 3, 1651–1662. [Google Scholar] [CrossRef]
- Oakley, G.G.; Tillison, K.; Opiyo, S.A.; Glanzer, J.G.; Horn, J.M.; Patrick, S.M. Physical interaction between replication protein A (RPA) and MRN: Involvement of RPA2 phosphorylation and the N-terminus of RPA1. Biochemistry 2009, 48, 7473–7481. [Google Scholar] [CrossRef]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef]
- Yamane, A.; Robbiani, D.F.; Resch, W.; Bothmer, A.; Nakahashi, H.; Oliveira, T.; Rommel, P.C.; Brown, E.J.; Nussenzweig, A.; Nussenzweig, M.C.; et al. RPA accumulation during class switch recombination represents 5′-3′ DNA-end resection during the S-G2/M phase of the cell cycle. Cell Rep. 2013, 3, 138–147. [Google Scholar] [CrossRef]
- Ziv, Y.; Bar-Shira, A.; Pecker, I.; Russell, P.; Jorgensen, T.J.; Tsarfati, I.; Shiloh, Y. Recombinant ATM protein complements the cellular A-T phenotype. Oncogene 1997, 15, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Ziv, Y.; Jaspers, N.G.; Etkin, S.; Danieli, T.; Trakhtenbrot, L.; Amiel, A.; Ravia, Y.; Shiloh, Y. Cellular and molecular characteristics of an immortalized ataxia- telangiectasia (group AB) cell line. Cancer Res. 1989, 49, 2495–2501. [Google Scholar] [PubMed]
- Guo, Z.; Kumagai, A.; Wang, S.X.; Dunphy, W.G. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 2000, 14, 2745–2756. [Google Scholar] [CrossRef]
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 2001, 21, 4129–4139. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.; Nievera, C.J.; Klimovich, V.; Fanning, E.; Wu, X. RPA2 is a direct downstream target for ATR to regulate the S-phase checkpoint. J. Biol. Chem. 2006, 281, 39517–39533. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Guntuku, S.; Cui, X.S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar] [CrossRef]
- Lindsey-Boltz, L.A.; Reardon, J.T.; Wold, M.S.; Sancar, A. In vitro analysis of the role of replication protein A (RPA) and RPA phosphorylation in ATR-mediated checkpoint signaling. J. Biol. Chem. 2012, 287, 36123–36131. [Google Scholar] [CrossRef]
- Liaw, H.; Lee, D.; Myung, K. DNA-PK-dependent RPA2 hyperphosphorylation facilitates DNA repair and suppresses sister chromatid exchange. PLoS ONE 2011, 6, e21424. [Google Scholar] [CrossRef]
- Anantha, R.W.; Sokolova, E.; Borowiec, J.A. RPA phosphorylation facilitates mitotic exit in response to mitotic DNA damage. Proc. Natl. Acad. Sci. USA 2008, 105, 12903–12908. [Google Scholar] [CrossRef] [PubMed]
- Carty, M.P.; Zernik-Kobak, M.; McGrath, S.; Dixon, K. UV light-induced DNA synthesis arrest in HeLa cells is associated with changes in phosphorylation of human single- stranded DNA-binding protein. EMBO J. 1994, 13, 2114–2123. [Google Scholar] [CrossRef] [PubMed]
- Stiff, T.; Cerosaletti, K.; Concannon, P.; O’Driscoll, M.; Jeggo, P.A. Replication independent ATR signalling leads to G2/M arrest requiring Nbs1, 53BP1 and MDC1. Hum. Mol. Genet. 2008, 17, 3247–3253. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, M.; Martinez-Pastor, B.; Murga, M.; Toledo, L.I.; Gutierrez-Martinez, P.; Lopez, E.; Fernandez-Capetillo, O. ATM regulates ATR chromatin loading in response to DNA double-strand breaks. J. Exp. Med. 2006, 203, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.S.; Cortez, D. Rapid activation of ATR by ionizing radiation requires ATM and Mre11. J. Biol. Chem. 2006, 281, 9346–9350. [Google Scholar] [CrossRef]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef]
- Beucher, A.; Birraux, J.; Tchouandong, L.; Barton, O.; Shibata, A.; Conrad, S.; Goodarzi, A.A.; Krempler, A.; Jeggo, P.A.; Lobrich, M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009, 28, 3413–3427. [Google Scholar] [CrossRef]
- Soniat, M.M.; Myler, L.R.; Paull, T.T.; Finkelstein, I.J. RPA phosphorylation regulates DNA resection. BioRxiv 2019, preprint. [Google Scholar]
- Lee, D.H.; Pan, Y.; Kanner, S.; Sung, P.; Borowiec, J.A.; Chowdhury, D. A PP4 phosphatase complex dephosphorylates RPA2 to facilitate DNA repair via homologous recombination. Nat. Struct. Mol. Biol. 2010, 17, 365–372. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Noon, A.T.; Deckbar, D.; Ziv, Y.; Shiloh, Y.; Lobrich, M.; Jeggo, P.A. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell 2008, 31, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Noon, A.T.; Shibata, A.; Rief, N.; Lobrich, M.; Stewart, G.S.; Jeggo, P.A.; Goodarzi, A.A. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat. Cell Biol. 2010, 12, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Ziv, Y.; Bielopolski, D.; Galanty, Y.; Lukas, C.; Taya, Y.; Schultz, D.C.; Lukas, J.; Bekker-Jensen, S.; Bartek, J.; Shiloh, Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol. 2006, 8, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Elia, A.E.; Wang, D.C.; Willis, N.A.; Boardman, A.P.; Hajdu, I.; Adeyemi, R.O.; Lowry, E.; Gygi, S.P.; Scully, R.; Elledge, S.J. RFWD3-Dependent Ubiquitination of RPA Regulates Repair at Stalled Replication Forks. Mol. Cell 2015, 60, 280–293. [Google Scholar] [CrossRef]
- Marechal, A.; Li, J.M.; Ji, X.Y.; Wu, C.S.; Yazinski, S.A.; Nguyen, H.D.; Liu, S.; Jimenez, A.E.; Jin, J.; Zou, L. PRP19 transforms into a sensor of RPA-ssDNA after DNA damage and drives ATR activation via a ubiquitin-mediated circuitry. Mol. Cell 2014, 53, 235–246. [Google Scholar] [CrossRef]
- Ashley, A.K.; Shrivastav, M.; Nie, J.; Amerin, C.; Troksa, K.; Glanzer, J.G.; Liu, S.; Opiyo, S.O.; Dimitrova, D.D.; Le, P.; et al. DNA-PK phosphorylation of RPA32 Ser4/Ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe. DNA Repair 2014, 21, 131–139. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Byrne, B.M.; Byrne, T.N.; Oakley, G.G. Role of RPA Phosphorylation in the ATR-Dependent G2 Cell Cycle Checkpoint. Genes 2023, 14, 2205. https://doi.org/10.3390/genes14122205
Liu S, Byrne BM, Byrne TN, Oakley GG. Role of RPA Phosphorylation in the ATR-Dependent G2 Cell Cycle Checkpoint. Genes. 2023; 14(12):2205. https://doi.org/10.3390/genes14122205
Chicago/Turabian StyleLiu, Shengqin, Brendan M. Byrne, Thomas N. Byrne, and Gregory G. Oakley. 2023. "Role of RPA Phosphorylation in the ATR-Dependent G2 Cell Cycle Checkpoint" Genes 14, no. 12: 2205. https://doi.org/10.3390/genes14122205