
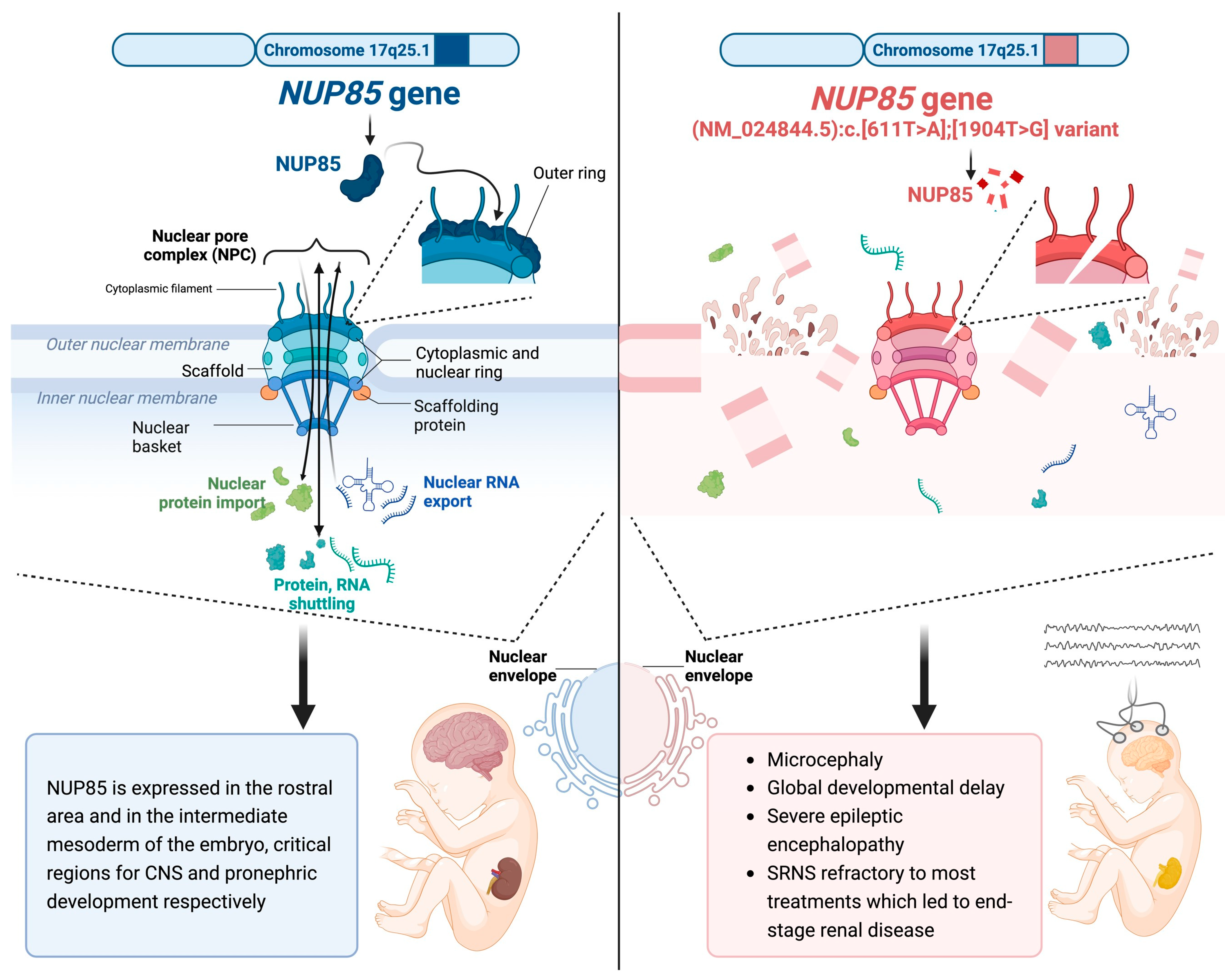
NUP85 as a Neurodevelopmental Gene: From Podocyte to Neuron
 , , , , , , , ,
, , , , , , , ,
Abstract
:1. Introduction
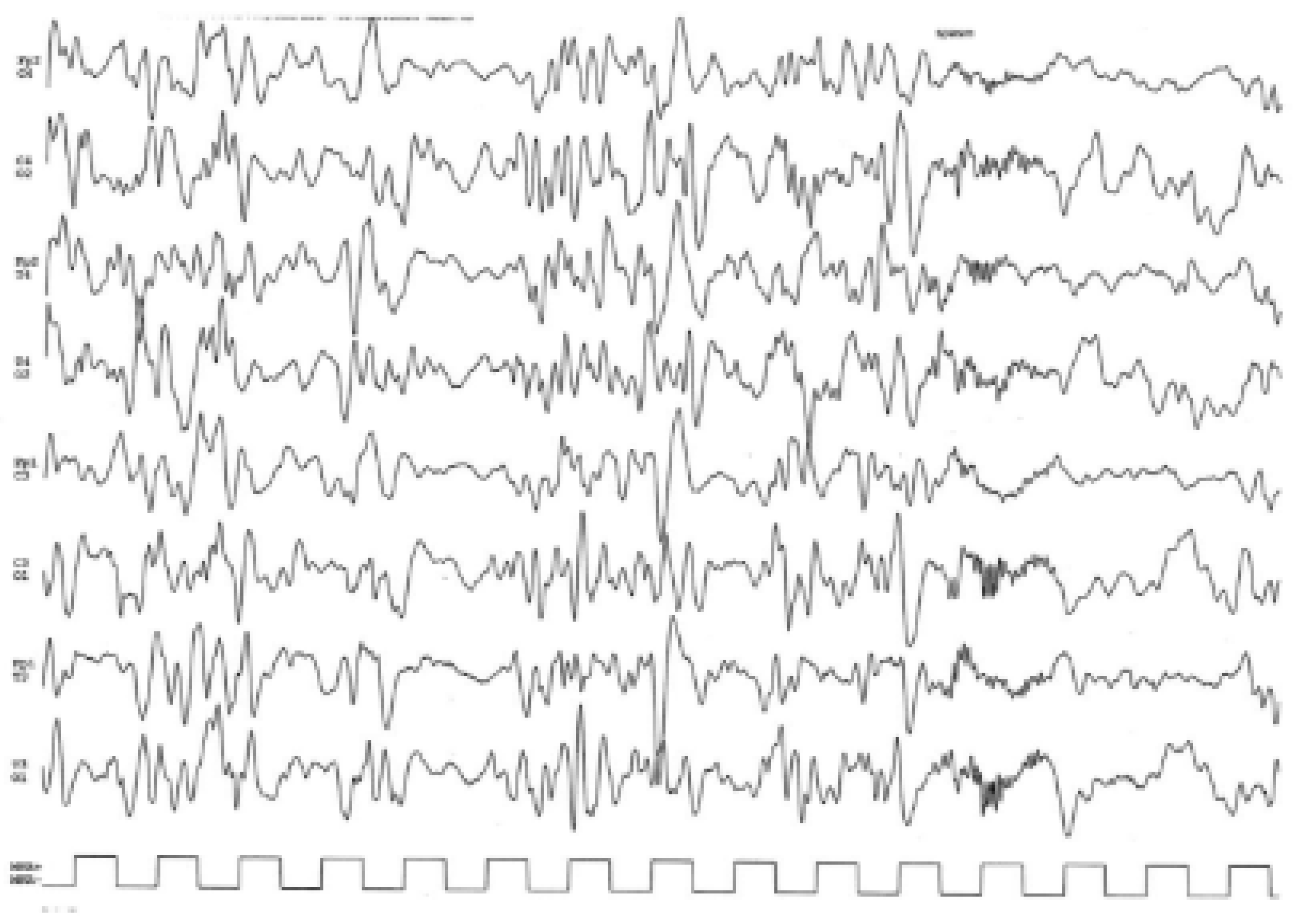
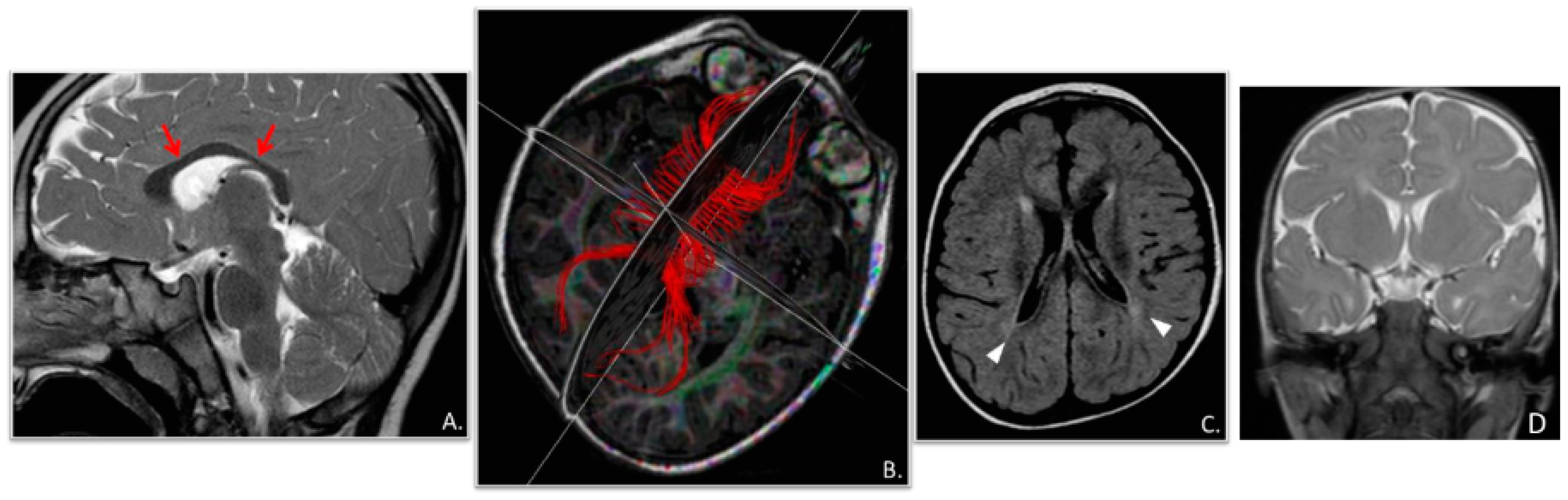
2. Clinical Report
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Downie, M.L.; Gallibois, C.; Parekh, R.S.; Noone, D.G. Nephrotic syndrome in infants and children: Pathophysiology and management. Paediatr. Int. Child Health 2017, 37, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, A.; Vivarelli, M.; Samuel, S.; Gipson, D.; Sinha, A.; Schaefer, F.; Hui, N.K.; Boyer, O.; Saleem, M.A.; Feltran, L.; et al. IPNA clinical practice recommendations for the diagnosis and management of children with steroid-resistant nephrotic syndrome. Pediatr. Nephrol. 2020, 35, 1529–1561. [Google Scholar] [CrossRef] [PubMed]
- Tullus, K.; Webb, H.; Bagga, A. Management of steroid-resistant nephrotic syndrome in children and adolescents. Lancet Child Adolesc. Health 2018, 2, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Iacomino, M.; Baldassari, S.; Tochigi, Y.; Kośla, K.; Buffelli, F.; Torella, A.; Severino, M.; Paladini, D.; Mandarà, L.; Riva, A.; et al. Loss of Wwox Perturbs Neuronal Migration and Impairs Early Cortical Development. Front. Neurosci. 2020, 14, 644. [Google Scholar] [CrossRef] [PubMed]
- Manole, A.; Efthymiou, S.; O’connor, E.; Mendes, M.I.; Jennings, M.; Maroofian, R.; Davagnanam, I.; Mankad, K.; Lopez, M.R.; Salpietro, V.; et al. De Novo and Bi-allelic Pathogenic Variants in NARS1 Cause Neurodevelopmental Delay Due to Toxic Gain-of-Function and Partial Loss-of-Function Effects. Am. J. Hum. Genet. 2020, 107, 311–324. [Google Scholar] [CrossRef]
- Dias, C.M.; Punetha, J.; Zheng, C.; Mazaheri, N.; Rad, A.; Efthymiou, S.; Petersen, A.; Dehghani, M.; Pehlivan, D.; Partlow, J.N.; et al. Homozygous Missense Variants in NTNG2, Encoding a Presynaptic Netrin-G2 Adhesion Protein, Lead to a Distinct Neurodevelopmental Disorder. Am. J. Hum. Genet. 2019, 105, 1048–1056. [Google Scholar] [CrossRef]
- Steel, D.; Salpietro, V.; Phadke, R.; Pitt, M.; Gentile, G.; Massoud, A.; Batten, L.; Bashamboo, A.; Mcelreavey, K.; Saggar, A.; et al. Whole exome sequencing reveals a MLL de novo mutation associated with mild developmental delay and without ‘hairy elbows’: Expanding the phenotype of Wiedemann–Steiner syndrome. J. Genet. 2015, 94, 755–758. [Google Scholar] [CrossRef]
- Jordan, P.; Dorval, G.; Arrondel, C.; Morinière, V.; Tournant, C.; Audrezet, M.; Michel-Calemard, L.; Putoux, A.; Lesca, G.; Labalme, A.; et al. Targeted next-generation sequencing in a large series of fetuses with severe renal diseases. Hum. Mutat. 2022, 43, 347–361. [Google Scholar] [CrossRef]
- Beck, M.; Hurt, E. The nuclear pore complex: Understanding its function through structural insight. Nat. Rev. Mol. Cell Biol. 2017, 18, 73–89. [Google Scholar] [CrossRef]
- Jühlen, R.; Fahrenkrog, B. Moonlighting nuclear pore proteins: Tissue-specific nucleoporin function in health and disease. Histochem. Cell Biol. 2018, 150, 593–605. [Google Scholar] [CrossRef]
- Guglielmi, V.; Sakuma, S.; D’Angelo, M.A. Nuclear pore complexes in development and tissue homeostasis. Development 2020, 147, dev183442. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, R.; Burke, B.; Doye, V. Nuclear transport and the mitotic apparatus: An evolving relationship. Cell. Mol. Life Sci. 2010, 67, 2215–2230. [Google Scholar] [CrossRef] [PubMed]
- Chatel, G.; Fahrenkrog, B. Nucleoporins: Leaving the nuclear pore complex for a successful mitosis. Cell. Signal. 2011, 23, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- del Viso, F.; Huang, F.; Myers, J.; Chalfant, M.; Zhang, Y.; Reza, N.; Bewersdorf, J.; Lusk, C.P.; Khokha, M.K. Congenital Heart Disease Genetics Uncovers Context-Dependent Organization and Function of Nucleoporins at Cilia. Dev. Cell 2016, 38, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Yang, S.; Han, Y.; Zhao, X.; Zhao, L.; Tian, T.; Tong, J.; Xu, P.; Xiong, C.; Meng, A. Loss of Zygotic NUP107 Protein Causes Missing of Pharyngeal Skeleton and Other Tissue Defects with Impaired Nuclear Pore Function in Zebrafish Embryos. J. Biol. Chem. 2012, 287, 38254–38264. [Google Scholar] [CrossRef]
- Lupu, F.; Alves, A.; Anderson, K.; Doye, V.; Lacy, E. Nuclear Pore Composition Regulates Neural Stem/Progenitor Cell Differentiation in the Mouse Embryo. Dev. Cell 2008, 14, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Reza, N.; Khokha, M.K.; Del Viso, F. Nucleoporin gene expression in Xenopus tropicalis embryonic development. Int. J. Dev. Biol. 2016, 60, 181–188. [Google Scholar] [CrossRef]
- Braun, D.A.; Lovric, S.; Schapiro, D.; Schneider, R.; Marquez, J.; Asif, M.; Hussain, M.S.; Daga, A.; Widmeier, E.; Rao, J.; et al. Mutations in multiple components of the nuclear pore complex cause nephrotic syndrome. J. Clin. Investig. 2018, 128, 4313–4328. [Google Scholar] [CrossRef]
- Ravindran, E.; Jühlen, R.; Vieira-Vieira, C.H.; Ha, T.; Salzberg, Y.; Fichtman, B.; Luise-Becker, L.; Martins, N.; Picker-Minh, S.; Bessa, P.; et al. Expanding the phenotype of NUP85 mutations beyond nephrotic syndrome to primary autosomal recessive microcephaly and Seckel syndrome spectrum disorders. Hum. Mol. Genet. 2021, 30, 2068–2081. [Google Scholar] [CrossRef]
- Niestroj, L.-M.; Perez-Palma, E.; Howrigan, D.P.; Zhou, Y.; Cheng, F.; Saarentaus, E.; Stevelink, R.; Daly, M.J.; Palotie, A.; Lal, D.; et al. Epilepsy subtype-specific copy number burden observed in a genome-wide study of 17 458 subjects. Brain 2020, 143, 2106–2118. [Google Scholar] [CrossRef]
- Wiessner, M.; Maroofian, R.; Ni, M.-Y.; Pedroni, A.; Müller, J.S.; Stucka, R.; Beetz, C.; Efthymiou, S.; Santorelli, F.M.; Alfares, A.; et al. Biallelic variants in HPDL cause pure and complicated hereditary spastic paraplegia. Brain 2021, 144, 1422–1434. [Google Scholar] [CrossRef] [PubMed]
- Dworschak, G.C.; Punetha, J.; Kalanithy, J.C.; Mingardo, E.; Erdem, H.B.; Akdemir, Z.C.; Karaca, E.; Mitani, T.; Marafi, D.; Fatih, J.M.; et al. Biallelic and monoallelic variants in PLXNA1 are implicated in a novel neurodevelopmental disorder with variable cerebral and eye anomalies. Anesthesia Analg. 2021, 23, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Donkervoort, S.; Kutzner, C.E.; Hu, Y.; Lornage, X.; Rendu, J.; Stojkovic, T.; Baets, J.; Neuhaus, S.B.; Tanboon, J.; Maroofian, R.; et al. Pathogenic Variants in the Myosin Chaperone UNC-45B Cause Progressive Myopathy with Eccentric Cores. Am. J. Hum. Genet. 2020, 107, 1078–1095. [Google Scholar] [CrossRef] [PubMed]
- Granata, F.; Morabito, R.; Mormina, E.; Alafaci, C.; Marino, S.; Laganà, A.; Vinci, S.L.; Briguglio, M.; Calamuneri, A.; Gaeta, M.; et al. 3T Double Inversion Recovery Magnetic Resonance Imaging: Diagnostic advantages in the evaluation of cortical development anomalies. Eur. J. Radiol. 2016, 85, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Colin, E.; Cong, E.H.; Mollet, G.; Guichet, A.; Gribouval, O.; Arrondel, C.; Boyer, O.; Daniel, L.; Gubler, M.-C.; Ekinci, Z.; et al. Loss-of-Function Mutations in WDR73 Are Responsible for Microcephaly and Steroid-Resistant Nephrotic Syndrome: Galloway-Mowat Syndrome. Am. J. Hum. Genet. 2014, 95, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Ben-Omran, T.; Fahiminiya, S.; Sorfazlian, N.; Almuriekhi, M.; Nawaz, Z.; Nadaf, J.; Abu Khadija, K.; Zaineddin, S.; Kamel, H.; Majewski, J.; et al. Nonsense mutation in the WDR73 gene is associated with Galloway-Mowat syndrome. J. Med. Genet. 2015, 52, 381–390. [Google Scholar] [CrossRef]
- Alazami, A.M.; Patel, N.; Shamseldin, H.E.; Anazi, S.; Al-Dosari, M.S.; Alzahrani, F.; Hijazi, H.; Alshammari, M.; Aldahmesh, M.A.; Salih, M.; et al. Accelerating Novel Candidate Gene Discovery in Neurogenetic Disorders via Whole-Exome Sequencing of Prescreened Multiplex Consanguineous Families. Cell Rep. 2015, 10, 148–161. [Google Scholar] [CrossRef]
- Rosti, R.O.; Sotak, B.N.; Bielas, S.L.; Bhat, G.; Silhavy, J.L.; Aslanger, A.D.; Altunoglu, U.; Bilge, I.; Tasdemir, M.; Yzaguirrem, A.D.; et al. Homozygous mutation in NUP107 leads to microcephaly with steroid-resistant nephrotic condition similar to Galloway-Mowat syndrome. J. Med. Genet. 2017, 54, 399–403. [Google Scholar] [CrossRef]
- Ravindran, E.; Lesca, G.; Januel, L.; Goldgruber, L.; Dickmanns, A.; Margot, H.; Kaindl, A.M. Case report: Compound heterozygous NUP85 variants cause autosomal recessive primary microcephaly. Front. Neurol. 2023, 14, 1124886. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Our Study | Ravindran et al., 2023 [29] | Ravindran et al., 2021 [19] | Braun et al., 2018 [18] | |||||
|---|---|---|---|---|---|---|---|---|
| Our Individual | Individual 1 | Individual 2 | Individual 3 | Individual 4 | Individual 5 | Individual 6 | Individual 7 | |
| NUP85 variant (NM_024844.5) | c.611T>A, c.1904T>G | c.454C>A, c.487C>A | c.932G>A | c.1109A>G, c.1589T>C | c.1430C>T | c.1933C>T | c.405+1G>A | c.1741G>C |
| Amino acid sequence changes | p.Val204Glu, p.Leu635Arg | p.Leu152Ile, p.Leu163Ile | p.Arg311Gln | p.Asn370Ser, p.Met530Thr | p.Ala477Val | p.Arg645Trp | Donor splice site | p.Ala581Pro |
| Parents’ consanguinity | − | − | + | − | + | − | − | − |
| Sex | Male | Male | Female | Female | Female | Male | Female | Male |
| Age at last assessment | 4 years | 3.6 years | 9 years | 27 GW | 8 years | 11 years | 7 years | 4 years |
| Age at onset | birth | birth | birth | prenatal | 8 years | 11 years | 7 years | 4 years |
| Congenital microcephaly | + | + | + | + | NC | NC | NC | NC |
| Intrauterine growth retardation | − | − | + | − | NC | NC | NC | NC |
| Short stature | − | + | + | − | + | − | − | + |
| Underweight | − | − | + | NC | NC | NC | NC | |
| Upslanted palpebral fissures | − | − | + | − | NC | NC | NC | NC |
| Short philtrum | − | + | + | − | NC | NC | NC | NC |
| High nasal bridge | − | − | + | − | NC | NC | NC | NC |
| Reduced vision | − | − | + | Unknown | NC | NC | NC | NC |
| Optic nerve atrophy | − | − | + | Unknown | NC | NC | NC | NC |
| Astigmatism | − | + | + | Unknown | NC | NC | NC | NC |
| Esophoria | − | + | + | Unknown | NC | NC | NC | NC |
| Long, skinny fingers | − | − | − | NC | NC | NC | NC | |
| Syndactyly | − | − | + | − | NC | NC | NC | NC |
| Pes adductus | − | + | + | − | NC | NC | NC | NC |
| Epilepsy | + | − | + | N/A | NC | NC | NC | NC |
| Intellectual disability, moderate | + | + | + | N/A | − | − | + | + |
| Delayed speech and language development | + | + | + | N/A | NC | NC | NC | NC |
| SRNS | + | − | − | N/A | + | + | + | + |
| Muscular hypotonia | + | + | + | N/A | NC | NC | NC | NC |
| Cranial MRI abnormalities | + | + | − | + | − | − | − | − |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambadauro, A.; Mangano, G.D.; Galletta, K.; Granata, F.; Riva, A.; Massella, L.; Guzzo, I.; Farello, G.; Scorrano, G.; Di Francesco, L.; et al. NUP85 as a Neurodevelopmental Gene: From Podocyte to Neuron. Genes 2023, 14, 2143. https://doi.org/10.3390/genes14122143
Gambadauro A, Mangano GD, Galletta K, Granata F, Riva A, Massella L, Guzzo I, Farello G, Scorrano G, Di Francesco L, et al. NUP85 as a Neurodevelopmental Gene: From Podocyte to Neuron. Genes. 2023; 14(12):2143. https://doi.org/10.3390/genes14122143
Chicago/Turabian StyleGambadauro, Antonella, Giuseppe Donato Mangano, Karol Galletta, Francesca Granata, Antonella Riva, Laura Massella, Isabella Guzzo, Giovanni Farello, Giovanna Scorrano, Ludovica Di Francesco, and et al. 2023. "NUP85 as a Neurodevelopmental Gene: From Podocyte to Neuron" Genes 14, no. 12: 2143. https://doi.org/10.3390/genes14122143