
Alternative Transcripts Diversify Genome Function for Phenome Relevance to Health and Diseases

Abstract
:1. Introduction
2. Alternative Exon Splicing (AES) Events: Features and Functions
2.1. AES—Increasingly Prevalent in Complex Organisms
2.2. AES—Dominantly Located in the Cytoplasm
2.3. AES—Commonly Expressed in Tissue-Specific Manner
2.4. AES—Highly Dysregulated in Neurological Diseases
3. Alternative Transcription Start (ATS) Events: Features and Functions
3.1. ATS—Genomically Aligned by Sequence Structures, Clustering Patterns, and Promoter Motifs
3.2. ATS—Highly Involved in Altered N-Terminal Proteins, Localization, Stability, and Complementary Functions
3.3. ATS—Frequently Tissue-Specific, Heavy Use of Intronic Enhancers
3.4. ATS—Commonly Linked with Tumor-Specific Oncogenesis, Invasion, and Metastasis
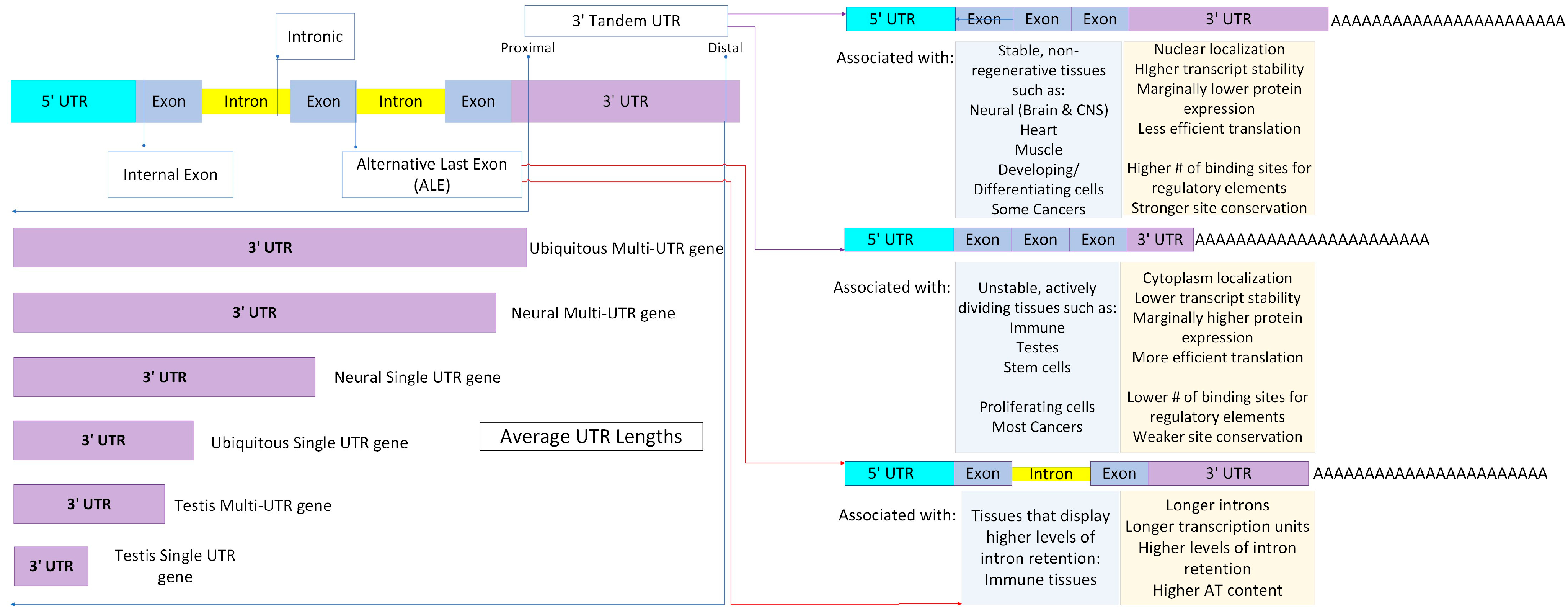
4. Alternative Polyadenylation (APA) Events: Features and Functions
4.1. APA—High Contribution to Transcript Diversity
4.2. APA—Involved in Transcript Stability and Translation Efficiency
4.3. APA—Tissue-Specific Processes in Response to Proliferation and Differentiation
4.4. APA—Significantly Associated with Diseases in High Differentiation or Proliferation Profiles
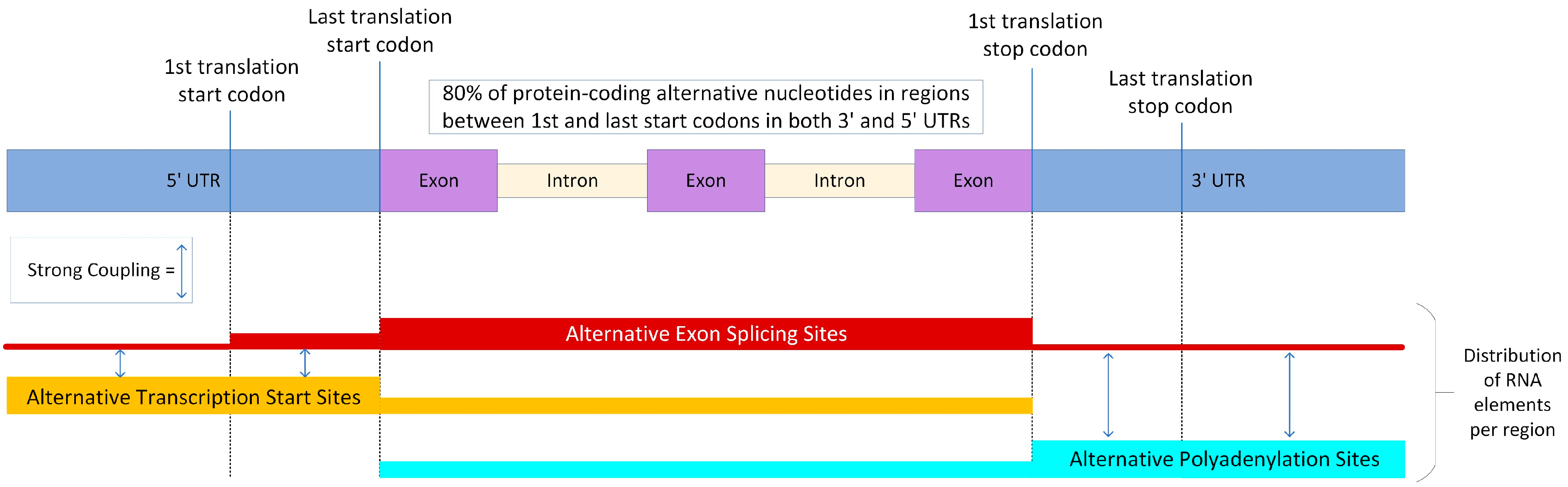
5. AES, ATS and APA Events: Evidence of Cooperation and Antagonism
6. Genome-Wide Profiling of RNA Variants: Challenges and Solutions
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Spuhler, J.N. On the Number of Genes in Man. Science 1948, 108, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Vogel, F. A Preliminary Estimate of the Number of Human Genes. Nature 1964, 201, 847. [Google Scholar] [CrossRef] [PubMed]
- Clancy, S. RNA Splicing: Introns, Exons and Spliceosome. Available online: http://www.nature.com/scitable/topicpage/rna-splicing-introns-exons-and-spliceosome-12375 (accessed on 2 January 2023).
- The Human Genome Project. Available online: https://www.genome.gov/human-genome-project (accessed on 2 January 2023).
- Understanding Our Genetic Inheritance: The US Human Genome Project, The First Five Years FY 1991–1995; National Center for Human Genome Research: Bethesda, MD, USA; USDOE Office of Energy Research: Washington, DC, USA; Office of Health and Environmental Research: Washington, DC, USA, 1990.
- Dunham, I.; Hunt, A.R.; Collins, J.E.; Bruskiewich, R.; Beare, D.M.; Clamp, M.; Smink, L.J.; Ainscough, R.; Almeida, J.P.; Babbage, A.; et al. The DNA Sequence of Human Chromosome 22. Nature 1999, 402, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Fields, C.; Adams, M.D.; White, O.; Venter, J.C. How Many Genes in the Human Genome? Nat. Genet. 1994, 7, 345–346. [Google Scholar] [CrossRef]
- Adams, M.D.; Kerlavage, A.R.; Fleischmann, R.D.; Fuldner, R.A.; Bult, C.J.; Lee, N.H.; Kirkness, E.F.; Weinstock, K.G.; Gocayne, J.D.; White, O. Initial Assessment of Human Gene Diversity and Expression Patterns Based upon 83 Million Nucleotides of cDNA Sequence. Nature 1995, 377, 3–174. [Google Scholar]
- Antequera, F.; Bird, A. Number of CpG Islands and Genes in Human and Mouse. Proc. Natl. Acad. Sci. USA 1993, 90, 11995–11999. [Google Scholar] [CrossRef]
- Liang, F.; Holt, I.; Pertea, G.; Karamycheva, S.; Salzberg, S.L.; Quackenbush, J. Gene Index Analysis of the Human Genome Estimates Approximately 120,000 Genes. Nat. Genet. 2000, 25, 239–240. [Google Scholar] [CrossRef]
- Wright, F.A.; Lemon, W.J.; Zhao, W.D.; Sears, R.; Zhuo, D.; Wang, J.-P.; Yang, H.-Y.; Baer, T.; Stredney, D.; Spitzner, J.; et al. A Draft Annotation and Overview of the Human Genome. Genome Biol. 2001, 2, 0025.1–0025.18. [Google Scholar] [CrossRef]
- Daly, M.J. Estimating the Human Gene Count. Cell 2002, 109, 283–284. [Google Scholar] [CrossRef]
- International Human Genome Sequencing Consortium. Finishing the Euchromatic Sequence of the Human Genome. Nature 2004, 431, 931–945. [Google Scholar] [CrossRef]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The Sequence of the Human Genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Juan, D.; Jungreis, I.; Martinez, L.; Rigau, M.; Rodriguez, J.M.; Vazquez, J.; Tress, M.L. Loose Ends: Almost One in Five Human Genes Still Have Unresolved Coding Status. Nucleic Acids Res. 2018, 46, 7070–7084. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The Complete Sequence of a Human Genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Zahler, A.M. Pre-mRNA Splicing and Its Regulation in Caenorhabditis Elegans; WormBook: Pasadena, CA, USA, 2018. [Google Scholar]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.M.; Pozo, F.; Cerdán-Vélez, D.; Di Domenico, T.; Vázquez, J.; Tress, M.L. APPRIS: Selecting Functionally Important Isoforms. Nucleic Acids Res. 2022, 50, D54–D59. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Huber, W. Alternative Start and Termination Sites of Transcription Drive Most Transcript Isoform Differences across Human Tissues. Nucleic Acids Res. 2018, 46, 582–592. [Google Scholar] [CrossRef]
- Shabalina, S.A.; Ogurtsov, A.Y.; Spiridonov, N.A.; Koonin, E.V. Evolution at Protein Ends: Major Contribution of Alternative Transcription Initiation and Termination to the Transcriptome and Proteome Diversity in Mammals. Nucleic Acids Res. 2014, 42, 7132–7144. [Google Scholar] [CrossRef]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Jungreis, I.; Lagarde, J.; Loveland, J.E.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Armstrong, J.; Barnes, I.; et al. GENCODE 2021. Nucleic Acids Res. 2021, 49, D916–D923. [Google Scholar] [CrossRef]
- Pertea, M.; Shumate, A.; Pertea, G.; Varabyou, A.; Breitwieser, F.P.; Chang, Y.-C.; Madugundu, A.K.; Pandey, A.; Salzberg, S.L. CHESS: A New Human Gene Catalog Curated from Thousands of Large-Scale RNA Sequencing Experiments Reveals Extensive Transcriptional Noise. Genome Biol. 2018, 19, 208. [Google Scholar] [CrossRef]
- Piovesan, A.; Antonaros, F.; Vitale, L.; Strippoli, P.; Pelleri, M.C.; Caracausi, M. Human Protein-Coding Genes and Gene Feature Statistics in 2019. BMC Res. Notes 2019, 12, 315. [Google Scholar] [CrossRef] [PubMed]
- Tress, M.L.; Abascal, F.; Valencia, A. Alternative Splicing May Not Be the Key to Proteome Complexity. Trends Biochem. Sci. 2017, 42, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Berget, S.M.; Moore, C.; Sharp, P.A. Spliced Segments at the 5’ Terminus of Adenovirus 2 Late mRNA. Proc. Natl. Acad. Sci. USA 1977, 74, 3171–3175. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, W. Why Genes in Pieces? Nature 1978, 271, 501. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Jiang, Y.; Zhong, M.; Wei, K.; Hu, C.; Qin, Y.; Zuo, Y.; Yang, L.; Shen, Z.; Zou, C. Regulatory Roles and Mechanisms of Alternative RNA Splicing in Adipogenesis and Human Metabolic Health. Cell Biosci. 2021, 11, 66. [Google Scholar] [CrossRef]
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [PubMed]
- Blencowe, B.J. Alternative Splicing: New Insights from Global Analyses. Cell 2006, 126, 37–47. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Huang, B.; Xu, Y.-M.; Li, J.; Huang, L.-F.; Lin, J.; Zhang, J.; Min, Q.-H.; Yang, W.-M.; et al. Mechanism of Alternative Splicing and Its Regulation (Review). Biomed. Rep. 2015, 3, 152–158. [Google Scholar] [CrossRef]
- Koren, E.; Lev-Maor, G.; Ast, G. The Emergence of Alternative 3′ and 5′ Splice Site Exons from Constitutive Exons. PLoS Comput. Biol. 2007, 3, e95. [Google Scholar] [CrossRef]
- Pozo, F.; Martinez-Gomez, L.; Walsh, T.A.; Rodriguez, J.M.; Di Domenico, T.; Abascal, F.; Vazquez, J.; Tress, M.L. Assessing the Functional Relevance of Splice Isoforms. NAR Genom. Bioinform. 2021, 3, lqab044. [Google Scholar] [CrossRef]
- Orr, M.W.; Mao, Y.; Storz, G.; Qian, S.-B. Alternative ORFs and Small ORFs: Shedding Light on the Dark Proteome. Nucleic Acids Res. 2020, 48, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Magny, E.G.; Pueyo, J.I.; Pearl, F.M.G.; Cespedes, M.A.; Niven, J.E.; Bishop, S.A.; Couso, J.P. Conserved Regulation of Cardiac Calcium Uptake by Peptides Encoded in Small Open Reading Frames. Science 2013, 341, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, Y.; Huang, Q.; Cheng, W.; Kang, Y.; Shu, L.; Yin, W.; Hua, Z.-C. Nuclear Localization of C-FLIP-L and Its Regulation of AP-1 Activity. Int. J. Biochem. Cell Biol. 2009, 41, 1678–1684. [Google Scholar] [CrossRef] [PubMed]
- Hillert, L.K.; Ivanisenko, N.V.; Espe, J.; König, C.; Ivanisenko, V.A.; Kähne, T.; Lavrik, I.N. Long and Short Isoforms of C-FLIP Act as Control Checkpoints of DED Filament Assembly. Oncogene 2020, 39, 1756–1772. [Google Scholar] [CrossRef]
- Qin, W.-S.; Wu, J.; Chen, Y.; Cui, F.-C.; Zhang, F.-M.; Lyu, G.-T.; Zhang, H.-M. The Short Isoform of Nuclear Mitotic Apparatus Protein 1 Functions as a Putative Tumor Suppressor. Chin. Med. J. (Engl.) 2017, 130, 1824–1830. [Google Scholar] [CrossRef]
- Identification and Characterization of Novel NuMA Isoforms. Biochem. Biophys. Res. Commun. 2014, 454, 387–392. [CrossRef]
- Bhuiyan, S.A.; Ly, S.; Phan, M.; Huntington, B.; Hogan, E.; Liu, C.C.; Liu, J.; Pavlidis, P. Systematic Evaluation of Isoform Function in Literature Reports of Alternative Splicing. BMC Genom. 2018, 19, 637. [Google Scholar] [CrossRef]
- Sangeeta Devi, Y.; Halperin, J. Reproductive Actions of Prolactin Mediated through Short and Long Receptor Isoforms. Mol. Cell. Endocrinol. 2014, 382, 400–410. [Google Scholar] [CrossRef]
- Bouilly, J.; Sonigo, C.; Auffret, J.; Gibori, G.; Binart, N. Prolactin Signaling Mechanisms in Ovary. Mol. Cell. Endocrinol. 2012, 356, 80–87. [Google Scholar] [CrossRef]
- Del Dotto, V.; Fogazza, M.; Carelli, V.; Rugolo, M.; Zanna, C. Eight Human OPA1 Isoforms, Long and Short: What Are They For? Biochim. Biophys. Acta BBA Bioenerg. 2018, 1859, 263–269. [Google Scholar] [CrossRef]
- Rodriguez, J.M.; Pozo, F.; di Domenico, T.; Vazquez, J.; Tress, M.L. An Analysis of Tissue-Specific Alternative Splicing at the Protein Level. PLoS Comput. Biol. 2020, 16, e1008287. [Google Scholar] [CrossRef] [PubMed]
- Baralle, F.E.; Giudice, J. Alternative Splicing as a Regulator of Development and Tissue Identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-H.; D, D.; Tarn, W.-Y. Alternative Splicing in Neurogenesis and Brain Development. Front. Mol. Biosci. 2018, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Vuong, C.K.; Black, D.L.; Zheng, S. The Neurogenetics of Alternative Splicing. Nat. Rev. Neurosci. 2016, 17, 265–281. [Google Scholar] [CrossRef] [PubMed]
- Nikonova, E.; Kao, S.-Y.; Spletter, M.L. Contributions of Alternative Splicing to Muscle Type Development and Function. Semin. Cell Dev. Biol. 2020, 104, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Gehring, N.H.; Roignant, J.-Y. Anything but Ordinary—Emerging Splicing Mechanisms in Eukaryotic Gene Regulation. Trends Genet. 2021, 37, 355–372. [Google Scholar] [CrossRef] [PubMed]
- Kalsotra, A.; Xiao, X.; Ward, A.J.; Castle, J.C.; Johnson, J.M.; Burge, C.B.; Cooper, T.A. A Postnatal Switch of CELF and MBNL Proteins Reprograms Alternative Splicing in the Developing Heart. Proc. Natl. Acad. Sci. USA 2008, 105, 20333–20338. [Google Scholar] [CrossRef] [PubMed]
- Naro, C.; Cesari, E.; Sette, C. Splicing Regulation in Brain and Testis: Common Themes for Highly Specialized Organs. Cell Cycle 2021, 20, 480–489. [Google Scholar] [CrossRef]
- Johnson, M.B.; Kawasawa, Y.I.; Mason, C.E.; Krsnik, Ž.; Coppola, G.; Bogdanović, D.; Geschwind, D.H.; Mane, S.M.; State, M.W.; Šestan, N. Functional and Evolutionary Insights into Human Brain Development through Global Transcriptome Analysis. Neuron 2009, 62, 494–509. [Google Scholar] [CrossRef]
- Ribba, A.-S.; Fraboulet, S.; Sadoul, K.; Lafanechère, L. The Role of LIM Kinases during Development: A Lens to Get a Glimpse of Their Implication in Pathologies. Cells 2022, 11, 403. [Google Scholar] [CrossRef]
- Jonkhout, N.; Tran, J.; Smith, M.A.; Schonrock, N.; Mattick, J.S.; Novoa, E.M. The RNA Modification Landscape in Human Disease. RNA 2017, 23, 1754–1769. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.A.; Wan, L.; Dreyfuss, G. RNA and Disease. Cell 2009, 136, 777–793. [Google Scholar] [CrossRef] [PubMed]
- Tazi, J.; Bakkour, N.; Stamm, S. Alternative Splicing and Disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2009, 1792, 14–26. [Google Scholar] [CrossRef]
- Shabalina, S.A.; Spiridonov, A.N.; Spiridonov, N.A.; Koonin, E.V. Connections between Alternative Transcription and Alternative Splicing in Mammals. Genome Biol. Evol. 2010, 2, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Nazim, M.; Masuda, A.; Rahman, M.A.; Nasrin, F.; Takeda, J.; Ohe, K.; Ohkawara, B.; Ito, M.; Ohno, K. Competitive Regulation of Alternative Splicing and Alternative Polyadenylation by hnRNP H and CstF64 Determines Acetylcholinesterase Isoforms. Nucleic Acids Res. 2017, 45, 1455–1468. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.M.; Swanson, M.S. RNA Mis-Splicing in Disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef]
- Davuluri, R.V.; Suzuki, Y.; Sugano, S.; Plass, C.; Huang, T.H.-M. The Functional Consequences of Alternative Promoter Use in Mammalian Genomes. Trends Genet. 2008, 24, 167–177. [Google Scholar] [CrossRef]
- Qin, Z.; Stoilov, P.; Zhang, X.; Xing, Y. SEASTAR: Systematic Evaluation of Alternative Transcription Start Sites in RNA. Nucleic Acids Res. 2018, 46, e45. [Google Scholar] [CrossRef]
- A Promoter-Level Mammalian Expression Atlas. Nature 2014, 507, 462–470. [CrossRef]
- Wang, X.; Hou, J.; Quedenau, C.; Chen, W. Pervasive Isoform-specific Translational Regulation via Alternative Transcription Start Sites in Mammals. Mol. Syst. Biol. 2016, 12, 875. [Google Scholar] [CrossRef]
- Lee, S.; Liu, B.; Lee, S.; Huang, S.-X.; Shen, B.; Qian, S.-B. Global Mapping of Translation Initiation Sites in Mammalian Cells at Single-Nucleotide Resolution. Proc. Natl. Acad. Sci. USA 2012, 109, E2424–E2432. [Google Scholar] [CrossRef] [PubMed]
- Benitez-Cantos, M.S.; Yordanova, M.M.; O’Connor, P.B.F.; Zhdanov, A.V.; Kovalchuk, S.I.; Papkovsky, D.B.; Andreev, D.E.; Baranov, P.V. Translation Initiation Downstream from Annotated Start Codons in Human mRNAs Coevolves with the Kozak Context. Genome Res. 2020, 30, 974–984. [Google Scholar] [CrossRef] [PubMed]
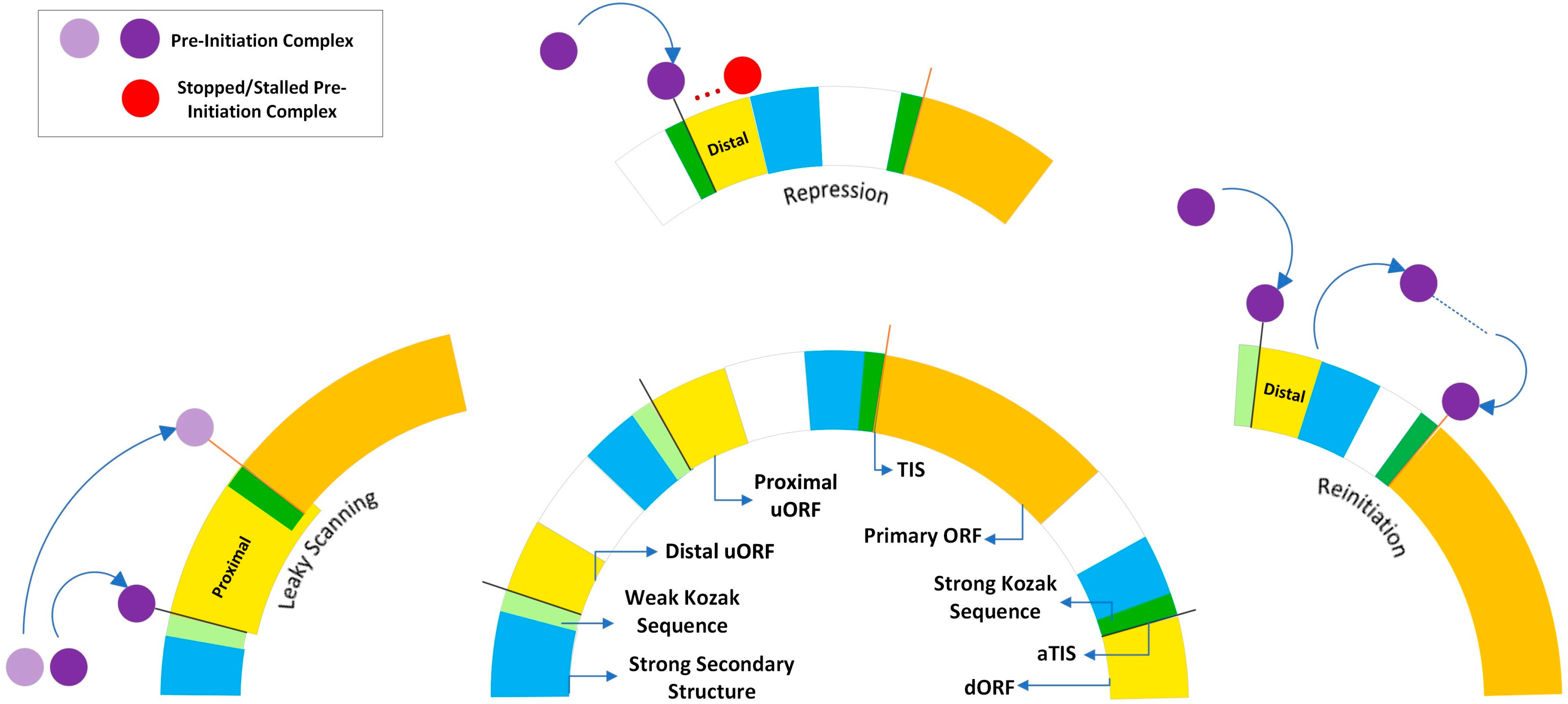
- Calvo, S.E.; Pagliarini, D.J.; Mootha, V.K. Upstream Open Reading Frames Cause Widespread Reduction of Protein Expression and Are Polymorphic among Humans. Proc. Natl. Acad. Sci. USA 2009, 106, 7507–7512. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, C.; Peixeiro, I.; Romão, L. Gene Expression Regulation by Upstream Open Reading Frames and Human Disease. PLoS Genet. 2013, 9, e1003529. [Google Scholar] [CrossRef] [PubMed]
- Trulley, P.; Snieckute, G.; Bekker-Jensen, D.; Menon, M.B.; Freund, R.; Kotlyarov, A.; Olsen, J.V.; Diaz-Muñoz, M.D.; Turner, M.; Bekker-Jensen, S.; et al. Alternative Translation Initiation Generates a Functionally Distinct Isoform of the Stress-Activated Protein Kinase MK2. Cell Rep. 2019, 27, 2859–2870.e6. [Google Scholar] [CrossRef]
- Liang, H.; Chen, X.; Yin, Q.; Ruan, D.; Zhao, X.; Zhang, C.; McNutt, M.A.; Yin, Y. PTENβ Is an Alternatively Translated Isoform of PTEN That Regulates rDNA Transcription. Nat. Commun. 2017, 8, 14771. [Google Scholar] [CrossRef]
- Murugan, M.; Fedele, D.; Millner, D.; Alharfoush, E.; Vegunta, G.; Boison, D. Adenosine Kinase: An Epigenetic Modulator in Development and Disease. Neurochem. Int. 2021, 147, 105054. [Google Scholar] [CrossRef]
- Lee, S.H.; Caviness, K.; Albright, E.R.; Lee, J.-H.; Gelbmann, C.B.; Rak, M.; Goodrum, F.; Kalejta, R.F. Long and Short Isoforms of the Human Cytomegalovirus UL138 Protein Silence IE Transcription and Promote Latency. J. Virol. 2016, 90, 9483–9494. [Google Scholar] [CrossRef]
- Demircioğlu, D.; Cukuroglu, E.; Kindermans, M.; Nandi, T.; Calabrese, C.; Fonseca, N.A.; Kahles, A.; Lehmann, K.-V.; Stegle, O.; Brazma, A.; et al. A Pan-Cancer Transcriptome Analysis Reveals Pervasive Regulation through Alternative Promoters. Cell 2019, 178, 1465–1477.e17. [Google Scholar] [CrossRef]
- Schug, J.; Schuller, W.-P.; Kappen, C.; Salbaum, J.M.; Bucan, M.; Stoeckert, C.J. Promoter Features Related to Tissue Specificity as Measured by Shannon Entropy. Genome Biol. 2005, 6, R33. [Google Scholar] [CrossRef]
- Jacox, E.; Gotea, V.; Ovcharenko, I.; Elnitski, L. Tissue-Specific and Ubiquitous Expression Patterns from Alternative Promoters of Human Genes. PLoS ONE 2010, 5, e12274. [Google Scholar] [CrossRef] [PubMed]
- Gacita, A.M.; Dellefave-Castillo, L.; Page, P.G.T.; Barefield, D.Y.; Wasserstrom, J.A.; Puckelwartz, M.J.; Nobrega, M.A.; McNally, E.M. Altered Enhancer and Promoter Usage Leads to Differential Gene Expression in the Normal and Failed Human Heart. Circ. Heart Fail. 2020, 13, e006926. [Google Scholar] [CrossRef] [PubMed]
- Floor, S.N.; Doudna, J.A. Tunable Protein Synthesis by Transcript Isoforms in Human Cells. eLife 2016, 5, e10921. [Google Scholar] [CrossRef] [PubMed]
- Karagyozov, L.; Godfrey, R.; Böhmer, S.-A.; Petermann, A.; Hölters, S.; Östman, A.; Böhmer, F.-D. The Structure of the 5′-End of the Protein-Tyrosine Phosphatase PTPRJ mRNA Reveals a Novel Mechanism for Translation Attenuation. Nucleic Acids Res. 2008, 36, 4443–4453. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Takizawa, T.; Takamori, K.; Ogawa, H.; Araki, Y. Genomic Organization and Structure of the 5′-Flanking Region of the TEX101 Gene: Alternative Promoter Usage and Splicing Generate Transcript Variants with Distinct 5′-Untranslated Region. Mol. Reprod. Dev. 2007, 74, 154–162. [Google Scholar] [CrossRef]
- Akman, B.H.; Can, T.; Erson-Bensan, A.E. Estrogen-Induced Upregulation and 3′-UTR Shortening of CDC6. Nucleic Acids Res. 2012, 40, 10679–10688. [Google Scholar] [CrossRef]
- Kiese, K.; Jablonski, J.; Boison, D.; Kobow, K. Dynamic Regulation of the Adenosine Kinase Gene during Early Postnatal Brain Development and Maturation. Front. Mol. Neurosci. 2016, 9, 99. [Google Scholar] [CrossRef]
- Liu, L.; Dilworth, D.; Gao, L.; Monzon, J.; Summers, A.; Lassam, N.; Hogg, D. Mutation of the CDKN2A 5’ UTR Creates an Aberrant Initiation Codon and Predisposes to Melanoma. Nat. Genet. 1999, 21, 128–132. [Google Scholar] [CrossRef]
- Sendoel, A.; Dunn, J.G.; Rodriguez, E.H.; Naik, S.; Gomez, N.C.; Hurwitz, B.; Levorse, J.; Dill, B.D.; Schramek, D.; Molina, H.; et al. Translation from Unconventional 5′ Start Sites Drives Tumour Initiation. Nature 2017, 541, 494–499. [Google Scholar] [CrossRef]
- Honda, S.; Arai, Y.; Haruta, M.; Sasaki, F.; Ohira, M.; Yamaoka, H.; Horie, H.; Nakagawara, A.; Hiyama, E.; Todo, S.; et al. Loss of Imprinting of IGF2 Correlates with Hypermethylation of the H19 Differentially Methylated Region in Hepatoblastoma. Br. J. Cancer 2008, 99, 1891–1899. [Google Scholar] [CrossRef]
- Dowdy, S.C.; Gostout, B.S.; Shridhar, V.; Wu, X.; Smith, D.I.; Podratz, K.C.; Jiang, S.-W. Biallelic Methylation and Silencing of Paternally Expressed Gene 3 (PEG3) in Gynecologic Cancer Cell Lines. Gynecol. Oncol. 2005, 99, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.; Early, P.; Carter, C.; Calame, K.; Bond, M.; Hood, L.; Wall, R. Two mRNAs with Different 3′ Ends Encode Membrane-Bound and Secreted Forms of Immunoglobulin μ Chain. Cell 1980, 20, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Setzer, D.R.; McGrogan, M.; Nunberg, J.H.; Schimke, R.T. Size Heterogeneity in the 3′ End of Dihydrofolate Reductase Messenger RNAs in Mouse Cells. Cell 1980, 22, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, L.; Qiu, Q.; Zhou, Q.; Ding, J.; Lu, Y.; Liu, P. Alternative Polyadenylation: Methods, Mechanism, Function, and Role in Cancer. J. Exp. Clin. Cancer Res. 2021, 40, 51. [Google Scholar] [CrossRef]
- Tang, P.; Yang, Y.; Li, G.; Huang, L.; Wen, M.; Ruan, W.; Guo, X.; Zhang, C.; Zuo, X.; Luo, D.; et al. Alternative Polyadenylation by Sequential Activation of Distal and Proximal PolyA Sites. Nat. Struct. Mol. Biol. 2022, 29, 21–31. [Google Scholar] [CrossRef]
- Derti, A.; Garrett-Engele, P.; MacIsaac, K.D.; Stevens, R.C.; Sriram, S.; Chen, R.; Rohl, C.A.; Johnson, J.M.; Babak, T. A Quantitative Atlas of Polyadenylation in Five Mammals. Genome Res. 2012, 22, 1173–1183. [Google Scholar] [CrossRef]
- Tian, B.; Manley, J.L. Alternative Polyadenylation of mRNA Precursors. Nat. Rev. Mol. Cell Biol. 2017, 18, 18–30. [Google Scholar] [CrossRef]
- Shi, Y. Alternative Polyadenylation: New Insights from Global Analyses. RNA 2012, 18, 2105–2117. [Google Scholar] [CrossRef]
- Hoque, M.; Ji, Z.; Zheng, D.; Luo, W.; Li, W.; You, B.; Park, J.Y.; Yehia, G.; Tian, B. Analysis of Alternative Cleavage and Polyadenylation by 3′ Region Extraction and Deep Sequencing. Nat. Methods 2013, 10, 133–139. [Google Scholar] [CrossRef]
- Evolution and Biological Roles of Alternative 3′UTRs. Trends Cell Biol. 2016, 26, 227–237. [CrossRef]
- Gruber, A.R.; Martin, G.; Müller, P.; Schmidt, A.; Gruber, A.J.; Gumienny, R.; Mittal, N.; Jayachandran, R.; Pieters, J.; Keller, W.; et al. Global 3′ UTR Shortening Has a Limited Effect on Protein Abundance in Proliferating T Cells. Nat. Commun. 2014, 5, 5465. [Google Scholar] [CrossRef]
- Spies, N.; Burge, C.B.; Bartel, D.P. 3′ UTR-Isoform Choice Has Limited Influence on the Stability and Translational Efficiency of Most mRNAs in Mouse Fibroblasts. Genome Res. 2013, 23, 2078–2090. [Google Scholar] [CrossRef] [PubMed]
- Geisberg, J.V.; Moqtaderi, Z.; Fan, X.; Ozsolak, F.; Struhl, K. Global Analysis of mRNA Isoform Half-Lives Reveals Stabilizing and Destabilizing Elements in Yeast. Cell 2014, 156, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Mittleman, B.E.; Pott, S.; Warland, S.; Zeng, T.; Mu, Z.; Kaur, M.; Gilad, Y.; Li, Y. Alternative Polyadenylation Mediates Genetic Regulation of Gene Expression. eLife 2020, 9, e57492. [Google Scholar] [CrossRef] [PubMed]
- Kakoki, M.; Pochynyuk, O.M.; Hathaway, C.M.; Tomita, H.; Hagaman, J.R.; Kim, H.-S.; Zaika, O.L.; Mamenko, M.; Kayashima, Y.; Matsuki, K.; et al. Primary Aldosteronism and Impaired Natriuresis in Mice Underexpressing TGFβ1. Proc. Natl. Acad. Sci. USA 2013, 110, 5600–5605. [Google Scholar] [CrossRef]
- Yeh, H.-S.; Yong, J. Alternative Polyadenylation of mRNAs: 3′-Untranslated Region Matters in Gene Expression. Mol. Cells 2016, 39, 281–285. [Google Scholar] [CrossRef]
- Agarwal, V.; Lopez-Darwin, S.; Kelley, D.R.; Shendure, J. The Landscape of Alternative Polyadenylation in Single Cells of the Developing Mouse Embryo. Nat. Commun. 2021, 12, 5101. [Google Scholar] [CrossRef]
- Xiao, R.; Li, C.; Wang, C.; Cao, Y.; Zhang, L.; Guo, Y.; Xin, Y.; Zhang, H.; Zhou, G. Adipogenesis Associated Mth938 Domain Containing (AAMDC) Protein Expression Is Regulated by Alternative Polyadenylation and microRNAs. FEBS Lett. 2019, 593, 1724–1734. [Google Scholar] [CrossRef]
- Bae, B.; Gruner, H.N.; Lynch, M.; Feng, T.; So, K.; Oliver, D.; Mastick, G.S.; Yan, W.; Pieraut, S.; Miura, P. Elimination of Calm1 Long 3′-UTR mRNA Isoform by CRISPR–Cas9 Gene Editing Impairs Dorsal Root Ganglion Development and Hippocampal Neuron Activation in Mice. RNA 2020, 26, 1414–1430. [Google Scholar] [CrossRef]
- Davis, A.G.; Einstein, J.M.; Zheng, D.; Jayne, N.D.; Fu, X.-D.; Tian, B.; Yeo, G.W.; Zhang, D.-E. A CRISPR RNA-Binding Protein Screen Reveals Regulators of RUNX1 Isoform Generation. Blood Adv. 2021, 5, 1310–1323. [Google Scholar] [CrossRef]
- GUAN, X.; FIERKE, C.A. Understanding Protein Palmitoylation: Biological Significance and Enzymology. Sci. China Chem. 2011, 54, 1888–1897. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Castro, I.; Garcia, B.C.; Curinha, A.; Neves-Costa, A.; Conde-Sousa, E.; Moita, L.F.; Moreira, A. MCL1 Alternative Polyadenylation Is Essential for Cell Survival and Mitochondria Morphology. Cell. Mol. Life Sci. 2022, 79, 164. [Google Scholar] [CrossRef] [PubMed]
- Andreassi, C.; Luisier, R.; Crerar, H.; Darsinou, M.; Blokzijl-Franke, S.; Lenn, T.; Luscombe, N.M.; Cuda, G.; Gaspari, M.; Saiardi, A.; et al. Cytoplasmic Cleavage of IMPA1 3′ UTR Is Necessary for Maintaining Axon Integrity. Cell Rep. 2021, 34, 108778. [Google Scholar] [CrossRef] [PubMed]
- Taliaferro, J.M.; Vidaki, M.; Oliveira, R.; Olson, S.; Zhan, L.; Saxena, T.; Wang, E.T.; Graveley, B.R.; Gertler, F.B.; Swanson, M.S.; et al. Distal Alternative Last Exons Localize mRNAs to Neural Projections. Mol. Cell 2016, 61, 821–833. [Google Scholar] [CrossRef]
- Lee, S.; Chen, Y.-C.; Gillen, A.E.; Taliaferro, J.M.; Deplancke, B.; Li, H.; Lai, E.C. Diverse Cell-Specific Patterns of Alternative Polyadenylation in Drosophila. Nat. Commun. 2022, 13, 5372. [Google Scholar] [CrossRef]
- Calame, K.L.; Lin, K.-I.; Tunyaplin, C. Regulatory Mechanisms That Determine the Development and Function of Plasma Cells. Annu. Rev. Immunol. 2003, 21, 205–230. [Google Scholar] [CrossRef]
- Galli, G.; Guise, J.W.; McDevitt, M.A.; Tucker, P.W.; Nevins, J.R. Relative Position and Strengths of Poly(A) Sites as Well as Transcription Termination Are Critical to Membrane versus Secreted Mu-Chain Expression during B-Cell Development. Genes Dev. 1987, 1, 471–481. [Google Scholar] [CrossRef]
- Singh, I.; Lee, S.-H.; Sperling, A.S.; Samur, M.K.; Tai, Y.-T.; Fulciniti, M.; Munshi, N.C.; Mayr, C.; Leslie, C.S. Widespread Intronic Polyadenylation Diversifies Immune Cell Transcriptomes. Nat. Commun. 2018, 9, 1716. [Google Scholar] [CrossRef]
- Gruber, A.J.; Zavolan, M. Alternative Cleavage and Polyadenylation in Health and Disease. Nat. Rev. Genet. 2019, 20, 599–614. [Google Scholar] [CrossRef]
- Curinha, A.; Oliveira Braz, S.; Pereira-Castro, I.; Cruz, A.; Moreira, A. Implications of Polyadenylation in Health and Disease. Nucleus 2014, 5, 508–519. [Google Scholar] [CrossRef]
- Rehfeld, A.; Plass, M.; Krogh, A.; Friis-Hansen, L. Alterations in Polyadenylation and Its Implications for Endocrine Disease. Front. Endocrinol. 2013, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- Anvar, S.Y.; Allard, G.; Tseng, E.; Sheynkman, G.M.; de Klerk, E.; Vermaat, M.; Yin, R.H.; Johansson, H.E.; Ariyurek, Y.; den Dunnen, J.T.; et al. Full-Length mRNA Sequencing Uncovers a Widespread Coupling between Transcription Initiation and mRNA Processing. Genome Biol. 2018, 19, 46. [Google Scholar] [CrossRef] [PubMed]
- Hsin, J.-P.; Manley, J.L. The RNA Polymerase II CTD Coordinates Transcription and RNA Processing. Genes Dev. 2012, 26, 2119–2137. [Google Scholar] [CrossRef] [PubMed]
- Batra, R.; Charizanis, K.; Manchanda, M.; Mohan, A.; Li, M.; Finn, D.J.; Goodwin, M.; Zhang, C.; Sobczak, K.; Thornton, C.A.; et al. Loss of MBNL Leads to Disruption of Developmentally Regulated Alternative Polyadenylation in RNA-Mediated Disease. Mol. Cell 2014, 56, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Kornblihtt, A.R.; Schor, I.E.; Alló, M.; Dujardin, G.; Petrillo, E.; Muñoz, M.J. Alternative Splicing: A Pivotal Step between Eukaryotic Transcription and Translation. Nat. Rev. Mol. Cell Biol. 2013, 14, 153–165. [Google Scholar] [CrossRef]
- Listerman, I.; Sapra, A.K.; Neugebauer, K.M. Cotranscriptional Coupling of Splicing Factor Recruitment and Precursor Messenger RNA Splicing in Mammalian Cells. Nat. Struct. Mol. Biol. 2006, 13, 815–822. [Google Scholar] [CrossRef]
- Babu, M.M. The Contribution of Intrinsically Disordered Regions to Protein Function, Cellular Complexity, and Human Disease. Biochem. Soc. Trans. 2016, 44, 1185–1200. [Google Scholar] [CrossRef]
- Schaarschmidt, S.; Fischer, A.; Zuther, E.; Hincha, D.K. Evaluation of Seven Different RNA-Seq Alignment Tools Based on Experimental Data from the Model Plant Arabidopsis Thaliana. Int. J. Mol. Sci. 2020, 21, 1720. [Google Scholar] [CrossRef]
- Corchete, L.A.; Rojas, E.A.; Alonso-López, D.; De Las Rivas, J.; Gutiérrez, N.C.; Burguillo, F.J. Systematic Comparison and Assessment of RNA-Seq Procedures for Gene Expression Quantitative Analysis. Sci. Rep. 2020, 10, 19737. [Google Scholar] [CrossRef]
- Robert, C.; Watson, M. Errors in RNA-Seq Quantification Affect Genes of Relevance to Human Disease. Genome Biol. 2015, 16, 177. [Google Scholar] [CrossRef]
- Assefa, A.T.; De Paepe, K.; Everaert, C.; Mestdagh, P.; Thas, O.; Vandesompele, J. Differential Gene Expression Analysis Tools Exhibit Substandard Performance for Long Non-Coding RNA-Sequencing Data. Genome Biol. 2018, 19, 96. [Google Scholar] [CrossRef] [PubMed]
- Zyprych-Walczak, J.; Szabelska, A.; Handschuh, L.; Górczak, K.; Klamecka, K.; Figlerowicz, M.; Siatkowski, I. The Impact of Normalization Methods on RNA-Seq Data Analysis. BioMed Res. Int. 2015, 2015, e621690. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Li, B.; Nelson, C.E.; Nabavi, S. Comparative Analysis of Differential Gene Expression Analysis Tools for Single-Cell RNA Sequencing Data. BMC Bioinform. 2019, 20, 40. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Reyes, A.; Huber, W. Detecting Differential Usage of Exons from RNA-Seq Data. Genome Res. 2012, 22, 2008–2017. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Mehmood, A.; Laiho, A.; Venäläinen, M.S.; McGlinchey, A.J.; Wang, N.; Elo, L.L. Systematic Evaluation of Differential Splicing Tools for RNA-Seq Studies. Brief. Bioinform. 2020, 21, 2052–2065. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Fenn, A.; Tsoy, O.; Faro, T.; Rössler, F.; Dietrich, A.; Kersting, J.; Louadi, Z.; Lio, C.T.; Völker, U.; Baumbach, J.; et al. Alternative Splicing Analysis Benchmark with DICAST. NAR Genom. Bioinform. 2022, 5, lqad044. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, S.; Yin, H.; Zhuo, Z.; Meng, G. A Comprehensive Benchmarking of Differential Splicing Tools for RNA-Seq Analysis at the Event Level. Brief. Bioinform. 2023, 24, bbad121. [Google Scholar] [CrossRef]
- Olofsson, D.; Preußner, M.; Kowar, A.; Heyd, F.; Neumann, A. One Pipeline to Predict Them All? On the Prediction of Alternative Splicing from RNA-Seq Data. Biochem. Biophys. Res. Commun. 2023, 653, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Park, J.W.; Lu, Z.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and Flexible Detection of Differential Alternative Splicing from Replicate RNA-Seq Data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [PubMed]
- Sterne-Weiler, T.; Weatheritt, R.J.; Best, A.; Ha, K.C.H.; Blencowe, B.J. Whippet: An Efficient Method for the Detection and Quantification of Alternative Splicing Reveals Extensive Transcriptomic Complexity. bioRxiv 2017. [Google Scholar] [CrossRef]
- Mancini, E.; Rabinovich, A.; Iserte, J.; Yanovsky, M.; Chernomoretz, A. ASpli: Integrative Analysis of Splicing Landscapes through RNA-Seq Assays. Bioinformatics 2021, 37, 2609–2616. [Google Scholar] [CrossRef]
- Goldstein, L.D.; Cao, Y.; Pau, G.; Lawrence, M.; Wu, T.D.; Seshagiri, S.; Gentleman, R. Prediction and Quantification of Splice Events from RNA-Seq Data. PLoS ONE 2016, 11, e0156132. [Google Scholar] [CrossRef]
- Middleton, R.; Gao, D.; Thomas, A.; Singh, B.; Au, A.; Wong, J.J.-L.; Bomane, A.; Cosson, B.; Eyras, E.; Rasko, J.E.J.; et al. IRFinder: Assessing the Impact of Intron Retention on Mammalian Gene Expression. Genome Biol. 2017, 18, 51. [Google Scholar] [CrossRef]
- Kahles, A.; Ong, C.S.; Zhong, Y.; Rätsch, G. SplAdder: Identification, Quantification and Testing of Alternative Splicing Events from RNA-Seq Data. Bioinformatics 2016, 32, 1840–1847. [Google Scholar] [CrossRef]
- Vaquero-Garcia, J.; Barrera, A.; Gazzara, M.R.; González-Vallinas, J.; Lahens, N.F.; Hogenesch, J.B.; Lynch, K.W.; Barash, Y. A New View of Transcriptome Complexity and Regulation through the Lens of Local Splicing Variations. eLife 2016, 5, e11752. [Google Scholar] [CrossRef]
- Huang, Y.; Sanguinetti, G. BRIE2: Computational Identification of Splicing Phenotypes from Single-Cell Transcriptomic Experiments. Genome Biol. 2021, 22, 251. [Google Scholar] [CrossRef]
- Thodberg, M.; Thieffry, A.; Vitting-Seerup, K.; Andersson, R.; Sandelin, A. CAGEfightR: Analysis of 5′-End Data Using R/Bioconductor. BMC Bioinform. 2019, 20, 487. [Google Scholar] [CrossRef]
- Policastro, R.A.; McDonald, D.J.; Brendel, V.P.; Zentner, G.E. Flexible Analysis of TSS Mapping Data and Detection of TSS Shifts with TSRexploreR. NAR Genom. Bioinform. 2021, 3, lqab051. [Google Scholar] [CrossRef] [PubMed]
- Cass, A.A.; Xiao, X. mountainClimber Identifies Alternative Transcription Start and Polyadenylation Sites in RNA-Seq. Cell Syst. 2019, 9, 393–400.e6. [Google Scholar] [CrossRef] [PubMed]
- Hou, R.; Hon, C.-C.; Huang, Y. CamoTSS: Analysis of Alternative Transcription Start Sites for Cellular Phenotypes and Regulatory Patterns from 5’ scRNA-Seq Data. bioRxiv 2023. [Google Scholar] [CrossRef]
- Shah, A.; Mittleman, B.E.; Gilad, Y.; Li, Y.I. Benchmarking Sequencing Methods and Tools That Facilitate the Study of Alternative Polyadenylation. Genome Biol. 2021, 22, 291. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Lian, Q.; Ye, C.; Wu, X. A Survey on Methods for Predicting Polyadenylation Sites from DNA Sequences, Bulk RNA-Seq, and Single-Cell RNA-Seq. Genom. Proteom. Bioinform. 2023, 21, 67–83. [Google Scholar] [CrossRef]
- Bryce-Smith, S.; Burri, D.; Gazzara, M.R.; Herrmann, C.J.; Danecka, W.; Fitzsimmons, C.M.; Wan, Y.K.; Zhuang, F.; Fansler, M.M.; Fernández, J.M.; et al. Extensible Benchmarking of Methods That Identify and Quantify Polyadenylation Sites from RNA-Seq Data. RNA, 2023; Online ahead of print. [Google Scholar] [CrossRef]
- Chen, M.; Ji, G.; Fu, H.; Lin, Q.; Ye, C.; Ye, W.; Su, Y.; Wu, X. A Survey on Identification and Quantification of Alternative Polyadenylation Sites from RNA-Seq Data. Brief. Bioinform. 2020, 21, 1261–1276. [Google Scholar] [CrossRef]
- Ye, C.; Long, Y.; Ji, G.; Li, Q.Q.; Wu, X. APAtrap: Identification and Quantification of Alternative Polyadenylation Sites from RNA-Seq Data. Bioinformatics 2018, 34, 1841–1849. [Google Scholar] [CrossRef]
- Arefeen, A.; Liu, J.; Xiao, X.; Jiang, T. TAPAS: Tool for Alternative Polyadenylation Site Analysis. Bioinformatics 2018, 34, 2521–2529. [Google Scholar] [CrossRef]
- Arefeen, A.; Xiao, X.; Jiang, T. DeepPASTA: Deep Neural Network Based Polyadenylation Site Analysis. Bioinformatics 2019, 35, 4577–4585. [Google Scholar] [CrossRef]
- Hao, J.; Kim, Y.; Kim, T.-K.; Kang, M. PASNet: Pathway-Associated Sparse Deep Neural Network for Prognosis Prediction from High-Throughput Data. BMC Bioinform. 2018, 19, 510. [Google Scholar] [CrossRef] [PubMed]
- Ji, G.; Zheng, J.; Shen, Y.; Wu, X.; Jiang, R.; Lin, Y.; Loke, J.C.; Davis, K.M.; Reese, G.J.; Li, Q.Q. Predictive Modeling of Plant Messenger RNA Polyadenylation Sites. BMC Bioinform. 2007, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Ha, K.C.H.; Blencowe, B.J.; Morris, Q. QAPA: A New Method for the Systematic Analysis of Alternative Polyadenylation from RNA-Seq Data. Genome Biol. 2018, 19, 45. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Donehower, L.A.; Cooper, T.A.; Neilson, J.R.; Wheeler, D.A.; Wagner, E.J.; Li, W. Dynamic Analyses of Alternative Polyadenylation from RNA-Seq Reveal a 3′-UTR Landscape across Seven Tumour Types. Nat. Commun. 2014, 5, 5274. [Google Scholar] [CrossRef]
- Wu, X.; Liu, T.; Ye, C.; Ye, W.; Ji, G. scAPAtrap: Identification and Quantification of Alternative Polyadenylation Sites from Single-Cell RNA-Seq Data. Brief. Bioinform. 2021, 22, bbaa273. [Google Scholar] [CrossRef]
- Shulman, E.D.; Elkon, R. Cell-Type-Specific Analysis of Alternative Polyadenylation Using Single-Cell Transcriptomics Data. Nucleic Acids Res. 2019, 47, 10027–10039. [Google Scholar] [CrossRef]
- Cheng, X.; Yan, J.; Liu, Y.; Wang, J.; Taubert, S. eVITTA: A Web-Based Visualization and Inference Toolbox for Transcriptome Analysis. Nucleic Acids Res. 2021, 49, W207–W215. [Google Scholar] [CrossRef]
- Flemington, E.K.; Flemington, S.A.; O’Grady, T.M.; Baddoo, M.; Nguyen, T.; Dong, Y.; Ungerleider, N.A. SpliceTools, a Suite of Downstream RNA Splicing Analysis Tools to Investigate Mechanisms and Impact of Alternative Splicing. Nucleic Acids Res. 2023, 51, e42. [Google Scholar] [CrossRef]
- Hu, X.; Song, J.; Chyr, J.; Wan, J.; Wang, X.; Du, J.; Duan, J.; Zhang, H.; Zhou, X.; Wu, X. APAview: A Web-Based Platform for Alternative Polyadenylation Analyses in Hematological Cancers. Front. Genet. 2022, 13, 928862. [Google Scholar] [CrossRef]
- Han, S.; Kim, D.; Kim, Y.; Choi, K.; Miller, J.E.; Kim, D.; Lee, Y. CAS-Viewer: Web-Based Tool for Splicing-Guided Integrative Analysis of Multi-Omics Cancer Data. BMC Med. Genom. 2018, 11, 25. [Google Scholar] [CrossRef]
- Hong, W.; Ruan, H.; Zhang, Z.; Ye, Y.; Liu, Y.; Li, S.; Jing, Y.; Zhang, H.; Diao, L.; Liang, H.; et al. APAatlas: Decoding Alternative Polyadenylation across Human Tissues. Nucleic Acids Res. 2020, 48, D34–D39. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, A.; Vural, S.; Zhao, Y.; Sonkin, D. MutSpliceDB: A Database of Splice Sites Variants with RNA-Seq Based Evidence on Effects on Splicing. Hum. Mutat. 2021, 42, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving Access to Variant Interpretations and Supporting Evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.; Yang, M.; Yiya, K.; Zhao, W.; Zhou, X. ExonSkipDB: Functional Annotation of Exon Skipping Event in Human. Nucleic Acids Res. 2020, 48, D896–D907. [Google Scholar] [CrossRef]
- Tapial, J.; Ha, K.C.H.; Sterne-Weiler, T.; Gohr, A.; Braunschweig, U.; Hermoso-Pulido, A.; Quesnel-Vallières, M.; Permanyer, J.; Sodaei, R.; Marquez, Y.; et al. An Atlas of Alternative Splicing Profiles and Functional Associations Reveals New Regulatory Programs and Genes That Simultaneously Express Multiple Major Isoforms. Genome Res. 2017, 27, 1759–1768. [Google Scholar] [CrossRef]
- Busch, A.; Hertel, K.J. HEXEvent: A Database of Human EXon Splicing Events. Nucleic Acids Res. 2013, 41, D118–D124. [Google Scholar] [CrossRef]
- Yamashita, R.; Sugano, S.; Suzuki, Y.; Nakai, K. DBTSS: DataBase of Transcriptional Start Sites Progress Report in 2012. Nucleic Acids Res. 2012, 40, D150–D154. [Google Scholar] [CrossRef]
- Abugessaisa, I.; Noguchi, S.; Hasegawa, A.; Kondo, A.; Kawaji, H.; Carninci, P.; Kasukawa, T. refTSS: A Reference Data Set for Human and Mouse Transcription Start Sites. J. Mol. Biol. 2019, 431, 2407–2422. [Google Scholar] [CrossRef]
- Herrmann, C.J.; Schmidt, R.; Kanitz, A.; Artimo, P.; Gruber, A.J.; Zavolan, M. PolyASite 2.0: A Consolidated Atlas of Polyadenylation Sites from 3′ End Sequencing. Nucleic Acids Res. 2020, 48, D174–D179. [Google Scholar] [CrossRef]
- Wang, R.; Nambiar, R.; Zheng, D.; Tian, B. PolyA_DB 3 Catalogs Cleavage and Polyadenylation Sites Identified by Deep Sequencing in Multiple Genomes. Nucleic Acids Res. 2018, 46, D315–D319. [Google Scholar] [CrossRef]
- Zhu, S.; Lian, Q.; Ye, W.; Qin, W.; Wu, Z.; Ji, G.; Wu, X. scAPAdb: A Comprehensive Database of Alternative Polyadenylation at Single-Cell Resolution. Nucleic Acids Res. 2022, 50, D365–D370. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, R.; Michal, J.J.; Wu, X.-L.; Liu, Z.; Zhao, H.; Xia, Y.; Du, W.; Wildung, M.R.; Pouchnik, D.J.; et al. Accurate Profiling of Gene Expression and Alternative Polyadenylation with Whole Transcriptome Termini Site Sequencing (WTTS-Seq). Genetics 2016, 203, 683–697. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhang, Y.; Michal, J.J.; Qu, L.; Zhang, S.; Wildung, M.R.; Du, W.; Pouchnik, D.J.; Zhao, H.; Xia, Y.; et al. Alternative Polyadenylation Coordinates Embryonic Development, Sexual Dimorphism and Longitudinal Growth in Xenopus Tropicalis. Cell. Mol. Life Sci. CMLS 2019, 76, 2185–2198. [Google Scholar] [CrossRef]
- Agrotis, A.; Pengo, N.; Burden, J.J.; Ketteler, R. Redundancy of Human ATG4 Protease Isoforms in Autophagy and LC3/GABARAP Processing Revealed in Cells. Autophagy 2019, 15, 976–997. [Google Scholar] [CrossRef]
- Foltran, R.B.; Diaz, S.L. BDNF Isoforms: A Round Trip Ticket between Neurogenesis and Serotonin? J. Neurochem. 2016, 138, 204–221. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Coksaygan, T.; Tang, H.; Khatri, R.; Balice-Gordon, R.J.; Rao, M.S.; Mattson, M.P. Truncated Tyrosine Kinase B Brain-Derived Neurotrophic Factor Receptor Directs Cortical Neural Stem Cells to a Glial Cell Fate by a Novel Signaling Mechanism. J. Neurochem. 2007, 100, 1515–1530. [Google Scholar] [CrossRef]
- Alsarraj, J.; Faraji, F.; Geiger, T.R.; Mattaini, K.R.; Williams, M.; Wu, J.; Ha, N.-H.; Merlino, T.; Walker, R.C.; Bosley, A.D.; et al. BRD4 Short Isoform Interacts with RRP1B, SIPA1 and Components of the LINC Complex at the Inner Face of the Nuclear Membrane. PLoS ONE 2013, 8, e80746. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Lee, C.-F.; Lai, H.-T.; Yu, C.-T.; Lee, J.-E.; Zuo, H.; Tsai, S.Y.; Tsai, M.-J.; Ge, K.; Wan, Y.; et al. Opposing Functions of BRD4 Isoforms in Breast Cancer. Mol. Cell 2020, 78, 1114–1132.e10. [Google Scholar] [CrossRef]
- Drumond-Bock, A.L.; Bieniasz, M. The Role of Distinct BRD4 Isoforms and Their Contribution to High-Grade Serous Ovarian Carcinoma Pathogenesis. Mol. Cancer 2021, 20, 145. [Google Scholar] [CrossRef]
- Bhattacherjee, A.; Jung, J.; Zia, S.; Ho, M.; Eskandari-Sedighi, G.; St. Laurent, C.D.; McCord, K.A.; Bains, A.; Sidhu, G.; Sarkar, S.; et al. The CD33 Short Isoform Is a Gain-of-Function Variant That Enhances Aβ1–42 Phagocytosis in Microglia. Mol. Neurodegener. 2021, 16, 19. [Google Scholar] [CrossRef]
- Ciolli Mattioli, C.; Rom, A.; Franke, V.; Imami, K.; Arrey, G.; Terne, M.; Woehler, A.; Akalin, A.; Ulitsky, I.; Chekulaeva, M. Alternative 3′ UTRs Direct Localization of Functionally Diverse Protein Isoforms in Neuronal Compartments. Nucleic Acids Res. 2019, 47, 2560–2573. [Google Scholar] [CrossRef] [PubMed]
- Phizicky, D.V.; Bell, S.P. Transcriptional Repression of CDC6 and SLD2 during Meiosis Is Associated with Production of Short Heterogeneous RNA Isoforms. Chromosoma 2018, 127, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, S.; Yokoyama, S.; Ueno, M.; Hayami, S.; Ieda, J.; Yamamoto, N.; Yamaguchi, S.; Mitani, Y.; Nakamura, Y.; Tani, M.; et al. CEACAM1 Long Cytoplasmic Domain Isoform Is Associated with Invasion and Recurrence of Hepatocellular Carcinoma. Ann. Surg. Oncol. 2014, 21, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Sadekova, S.; Lamarche-Vane, N.; Li, X.; Beauchemin, N. The CEACAM1-L Glycoprotein Associates with the Actin Cytoskeleton and Localizes to Cell–Cell Contact through Activation of Rho-like GTPases. Mol. Biol. Cell 2000, 11, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, F.; Barberi, L.; Dobrowolny, G.; Villa Nova Bacurau, A.; Nicoletti, C.; Rizzuto, E.; Rosenthal, N.; Scicchitano, B.M.; Musarò, A. Effects of IGF-1 Isoforms on Muscle Growth and Sarcopenia. Aging Cell 2019, 18, e12954. [Google Scholar] [CrossRef]
- Annibalini, G.; Contarelli, S.; De Santi, M.; Saltarelli, R.; Di Patria, L.; Guescini, M.; Villarini, A.; Brandi, G.; Stocchi, V.; Barbieri, E. The Intrinsically Disordered E-Domains Regulate the IGF-1 Prohormones Stability, Subcellular Localisation and Secretion. Sci. Rep. 2018, 8, 8919. [Google Scholar] [CrossRef]
- Philippou, A.; Maridaki, M.; Pneumaticos, S.; Koutsilieris, M. The Complexity of the IGF1 Gene Splicing, Posttranslational Modification and Bioactivity. Mol. Med. 2014, 20, 202–214. [Google Scholar] [CrossRef]
- Lewis-Tuffin, L.J.; Jewell, C.M.; Bienstock, R.J.; Collins, J.B.; Cidlowski, J.A. Human Glucocorticoid Receptor β Binds RU-486 and Is Transcriptionally Active. Mol. Cell. Biol. 2007, 27, 2266–2282. [Google Scholar] [CrossRef]
- Dierolf, J.G.; Watson, A.J.; Betts, D.H. Differential Localization Patterns of Pyruvate Kinase Isoforms in Murine Naïve, Formative, and Primed Pluripotent States. Exp. Cell Res. 2021, 405, 112714. [Google Scholar] [CrossRef]
- Taniguchi, K.; Ito, Y.; Sugito, N.; Kumazaki, M.; Shinohara, H.; Yamada, N.; Nakagawa, Y.; Sugiyama, T.; Futamura, M.; Otsuki, Y.; et al. Organ-Specific PTB1-Associated microRNAs Determine Expression of Pyruvate Kinase Isoforms. Sci. Rep. 2015, 5, 8647. [Google Scholar] [CrossRef]
- Kiviluoto, S.; Decuypere, J.-P.; De Smedt, H.; Missiaen, L.; Parys, J.B.; Bultynck, G. STIM1 as a Key Regulator for Ca2+ Homeostasis in Skeletal-Muscle Development and Function. Skelet. Muscle 2011, 1, 16. [Google Scholar] [CrossRef] [PubMed]
- Knapp, M.L.; Alansary, D.; Poth, V.; Förderer, K.; Sommer, F.; Zimmer, D.; Schwarz, Y.; Künzel, N.; Kless, A.; Machaca, K.; et al. A Longer Isoform of Stim1 Is a Negative SOCE Regulator but Increases cAMP-Modulated NFAT Signaling. EMBO Rep. 2022, 23, e53135. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G.; Jarzembowski, L.; Schwarz, Y.; Poth, V.; Konrad, M.; Knapp, M.L.; Schwär, G.; Lauer, A.A.; Grimm, M.O.W.; Alansary, D.; et al. A Short Isoform of STIM1 Confers Frequency-Dependent Synaptic Enhancement. Cell Rep. 2021, 34, 108844. [Google Scholar] [CrossRef]
- Goldsmith, J.F.; Hall, C.G.; Atkinson, T.P. Identification of an Alternatively Spliced Isoform of the Fyn Tyrosine Kinase. Biochem. Biophys. Res. Commun. 2002, 298, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Brignatz, C.; Paronetto, M.P.; Opi, S.; Cappellari, M.; Audebert, S.; Feuillet, V.; Bismuth, G.; Roche, S.; Arold, S.T.; Sette, C.; et al. Alternative Splicing Modulates Autoinhibition and SH3 Accessibility in the Src Kinase Fyn. Mol. Cell. Biol. 2009, 29, 6438–6448. [Google Scholar] [CrossRef] [PubMed]
- Picard, C.; Gabert, J.; Olive, D.; Collette, Y. Altered Splicing in Hematological Malignancies Reveals a Tissue-Specific Translational Block of the Src-Family Tyrosine Kinase Fyn Brain Isoform Expression. Leukemia 2004, 18, 1737–1739. [Google Scholar] [CrossRef]
- Toutant, M.; Costa, A.; Studler, J.-M.; Kadaré, G.; Carnaud, M.; Girault, J.-A. Alternative Splicing Controls the Mechanisms of FAK Autophosphorylation. Mol. Cell. Biol. 2002, 22, 7731–7743. [Google Scholar] [CrossRef]
- Jereb, S.; Hwang, H.-W.; Van Otterloo, E.; Govek, E.-E.; Fak, J.J.; Yuan, Y.; Hatten, M.E.; Darnell, R.B. Differential 3’ Processing of Specific Transcripts Expands Regulatory and Protein Diversity across Neuronal Cell Types. eLife 2018, 7, e34042. [Google Scholar] [CrossRef]
- Wang, J.-Z.; Fu, X.; Fang, Z.; Liu, H.; Zong, F.-Y.; Zhu, H.; Yu, Y.-F.; Zhang, X.-Y.; Wang, S.-F.; Huang, Y.; et al. QKI-5 Regulates the Alternative Splicing of Cytoskeletal Gene ADD3 in Lung Cancer. J. Mol. Cell Biol. 2020, 13, 347–360. [Google Scholar] [CrossRef]
- Wang, Y.; Vogel, G.; Yu, Z.; Richard, S. The QKI-5 and QKI-6 RNA Binding Proteins Regulate the Expression of MicroRNA 7 in Glial Cells. Mol. Cell. Biol. 2013, 33, 1233–1243. [Google Scholar] [CrossRef]
- Larocque, D.; Fragoso, G.; Huang, J.; Mushynski, W.E.; Loignon, M.; Richard, S.; Almazan, G. The QKI-6 and QKI-7 RNA Binding Proteins Block Proliferation and Promote Schwann Cell Myelination. PLoS ONE 2009, 4, e5867. [Google Scholar] [CrossRef] [PubMed]
- Pattwell, S.S.; Arora, S.; Cimino, P.J.; Ozawa, T.; Szulzewsky, F.; Hoellerbauer, P.; Bonifert, T.; Hoffstrom, B.G.; Boiani, N.E.; Bolouri, H.; et al. A Kinase-Deficient NTRK2 Splice Variant Predominates in Glioma and Amplifies Several Oncogenic Signaling Pathways. Nat. Commun. 2020, 11, 2977. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Hammond, E.; Rabbitts, P. Temporal and Spatial Expression of Two Isoforms of the Dutt1/Robo1 Gene in Mouse Development. FEBS Lett. 2002, 523, 12–16. [Google Scholar] [CrossRef]
- Camurri, L.; Mambetisaeva, E.; Davies, D.; Parnavelas, J.; Sundaresan, V.; Andrews, W. Evidence for the Existence of Two Robo3 Isoforms with Divergent Biochemical Properties. Mol. Cell. Neurosci. 2005, 30, 485–493. [Google Scholar] [CrossRef]
- Ruedel, A.; Schott, M.; Schubert, T.; Bosserhoff, A.K. Robo3A and Robo3B Expression Is Regulated via Alternative Promoters and mRNA Stability. Cancer Cell Int. 2016, 16, 71. [Google Scholar] [CrossRef] [PubMed]
- Ikebe, C.; Ohashi, K.; Fujimori, T.; Bernard, O.; Noda, T.; Robertson, E.J.; Mizuno, K. Mouse LIM-Kinase 2 Gene: cDNA Cloning, Genomic Organization, and Tissue-Specific Expression of Two Alternatively Initiated Transcripts. Genomics 1997, 46, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Subcellular Localization and Protein Interaction of the Human LIMK2 Gene Expressing Alternative Transcripts with Tissue-Specific Regulation. Biochem. Biophys. Res. Commun. 1996, 229, 582–589. [CrossRef]
- Ahn, J.-Y.; Rong, R.; Kroll, T.G.; Meir, E.G.V.; Snyder, S.H.; Ye, K. PIKE (Phosphatidylinositol 3-Kinase Enhancer)-A GTPase Stimulates Akt Activity and Mediates Cellular Invasion*. J. Biol. Chem. 2004, 279, 16441–16451. [Google Scholar] [CrossRef]
- Ahn, J.-Y.; Ye, K. PIKE GTPase Signaling and Function. Int. J. Biol. Sci. 2005, 1, 44–50. [Google Scholar] [CrossRef]
- Han, C.; Zhao, R.; Kroger, J.; Qu, M.; Wani, A.A.; Wang, Q.-E. Caspase-2 Short Isoform Interacts with Membrane-Associated Cytoskeleton Proteins to Inhibit Apoptosis. PLoS ONE 2013, 8, e67033. [Google Scholar] [CrossRef]
- Logette, E.; Wotawa, A.; Solier, S.; Desoche, L.; Solary, E.; Corcos, L. The Human Caspase-2 Gene: Alternative Promoters, Pre-mRNA Splicing and AUG Usage Direct Isoform-Specific Expression. Oncogene 2003, 22, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Benassayag, C.; Montero, L.; Colombié, N.; Gallant, P.; Cribbs, D.; Morello, D. Human C-Myc Isoforms Differentially Regulate Cell Growth and Apoptosis in Drosophila Melanogaster. Mol. Cell. Biol. 2005, 25, 9897–9909. [Google Scholar] [CrossRef] [PubMed]
- Diernfellner, A.; Colot, H.V.; Dintsis, O.; Loros, J.J.; Dunlap, J.C.; Brunner, M. Long and Short Isoforms of Neurospora Clock Protein FRQ Support Temperature Compensated Circadian Rhythms. FEBS Lett. 2007, 581, 5759–5764. [Google Scholar] [CrossRef]
- Cha, J.; Yuan, H.; Liu, Y. Regulation of the Activity and Cellular Localization of the Circadian Clock Protein FRQ. J. Biol. Chem. 2011, 286, 11469–11478. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, S.W.; Gauthier, A.E.; Mills, E.W.; Ingolia, N.T.; Kagan, J.C. A Bicistronic MAVS Transcript Highlights a Class of Truncated Variants in Antiviral Immunity. Cell 2014, 156, 800–811. [Google Scholar] [CrossRef]
- Vazquez, C.; Beachboard, D.C.; Horner, S.M. Methods to Visualize MAVS Subcellular Localization. Methods Mol. Biol. 2017, 1656, 131–142. [Google Scholar] [CrossRef]
- Qi, N.; Shi, Y.; Zhang, R.; Zhu, W.; Yuan, B.; Li, X.; Wang, C.; Zhang, X.; Hou, F. Multiple Truncated Isoforms of MAVS Prevent Its Spontaneous Aggregation in Antiviral Innate Immune Signalling. Nat. Commun. 2017, 8, 15676. [Google Scholar] [CrossRef] [PubMed]
- Kadmiel, M.; Cidlowski, J.A. Glucocorticoid Receptor Signaling in Health and Disease. Trends Pharmacol. Sci. 2013, 34, 518–530. [Google Scholar] [CrossRef]
- Oakley, R.H.; Cidlowski, J.A. Cellular Processing of the Glucocorticoid Receptor Gene and Protein: New Mechanisms for Generating Tissue-Specific Actions of Glucocorticoids. J. Biol. Chem. 2011, 286, 3177–3184. [Google Scholar] [CrossRef]
- Jenkins, A.K.; Paterson, C.; Wang, Y.; Hyde, T.M.; Kleinman, J.E.; Law, A.J. Neurexin 1 (NRXN1) Splice Isoform Expression During Human Neocortical Development and Aging. Mol. Psychiatry 2016, 21, 701–706. [Google Scholar] [CrossRef]
- Vieler, M.; Sanyal, S. P53 Isoforms and Their Implications in Cancer. Cancers 2018, 10, 288. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.-C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. P53 Isoforms Can Regulate P53 Transcriptional Activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef] [PubMed]
- Khoury, M.P.; Bourdon, J.-C. The Isoforms of the P53 Protein. Cold Spring Harb. Perspect. Biol. 2010, 2, a000927. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.-Y.; Whitfield, P.; Llorian, M.; Monzon-Casanova, E.; Diaz-Munoz, M.D.; Turner, M.; Smith, C.W.J. Generation of Functionally Distinct Isoforms of PTBP3 by Alternative Splicing and Translation Initiation. Nucleic Acids Res. 2015, 43, 5586–5600. [Google Scholar] [CrossRef]
- Huang, S.N.; Dalla Rosa, I.; Michaels, S.A.; Tulumello, D.V.; Agama, K.; Khiati, S.; Jean, S.R.; Baechler, S.A.; Factor, V.M.; Varma, S.; et al. Mitochondrial Tyrosyl-DNA Phosphodiesterase 2 and Its TDP2S Short Isoform. EMBO Rep. 2018, 19, e42139. [Google Scholar] [CrossRef]
- Graham, R.R.; Kyogoku, C.; Sigurdsson, S.; Vlasova, I.A.; Davies, L.R.L.; Baechler, E.C.; Plenge, R.M.; Koeuth, T.; Ortmann, W.A.; Hom, G.; et al. Three Functional Variants of IFN Regulatory Factor 5 (IRF5) Define Risk and Protective Haplotypes for Human Lupus. Proc. Natl. Acad. Sci. USA 2007, 104, 6758–6763. [Google Scholar] [CrossRef]
- Abbas, W.; Kumar, A.; Herbein, G. The eEF1A Proteins: At the Crossroads of Oncogenesis, Apoptosis, and Viral Infections. Front. Oncol. 2015, 5, 75. [Google Scholar] [CrossRef]
- Manzo, M.; Wirz, J.; Ambrosi, C.; Villaseñor, R.; Roschitzki, B.; Baubec, T. Isoform-Specific Localization of DNMT3A Regulates DNA Methylation Fidelity at Bivalent CpG Islands. EMBO J. 2017, 36, 3421–3434. [Google Scholar] [CrossRef]
- Lax, E.; Sapozhnikov, D.M. Dnmt3a2 in the Nucleus Accumbens Shell Mediates Cue-Induced Cocaine-Seeking Behavior. J. Neurosci. 2019, 39, 2574–2576. [Google Scholar] [CrossRef]
- Zhang, Z.; So, K.; Peterson, R.; Bauer, M.; Ng, H.; Zhang, Y.; Kim, J.H.; Kidd, T.; Miura, P. Elav-Mediated Exon Skipping and Alternative Polyadenylation of the Dscam1 Gene Are Required for Axon Outgrowth. Cell Rep. 2019, 27, 3808–3817.e7. [Google Scholar] [CrossRef]
- Jeon, S.; Kim, Y.; Jeong, Y.M.; Bae, J.S.; Jung, C.K. CCND1 Splice Variant as A Novel Diagnostic and Predictive Biomarker for Thyroid Cancer. Cancers 2018, 10, 437. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; He, G.; Hou, M.; Chen, L.; Chen, S.; Xu, A.; Fu, Y. Cell Cycle Regulation by Alternative Polyadenylation of CCND1. Sci. Rep. 2018, 8, 6824. [Google Scholar] [CrossRef] [PubMed]
- Carr, H.S.; Morris, C.A.; Menon, S.; Song, E.H.; Frost, J.A. Rac1 Controls the Subcellular Localization of the Rho Guanine Nucleotide Exchange Factor Net1A To Regulate Focal Adhesion Formation and Cell Spreading. Mol. Cell. Biol. 2013, 33, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Winter, C.; Pawel, B.; Seiser, E.; Zhao, H.; Raabe, E.; Wang, Q.; Judkins, A.R.; Attiyeh, E.; Maris, J.M. Neural cell adhesion molecule (NCAM) isoform expression is associated with neuroblastoma differentiation status. Pediatr. Blood Cancer 2008, 51, 10–16. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Method Type | Method Name | Program Environment | Introductory Article |
|---|---|---|---|
| Recommended DE Tools | limma | Bioconductor R package | https://doi.org/10.1093/nar/gkv007, accessed on 1 November 2023 |
| edgeR | Bioconductor R package | https://doi.org/10.1093/bioinformatics/btp616, accessed on 1 November 2023 | |
| DESeq2 | Bioconductor R package | https://doi.org/10.1186/s13059-014-0550-8, accessed on 1 November 2023 | |
| Recommended AES Tools | rMATs | R package | https://rnaseq-mats.sourceforge.io/, accessed on 1 November 2023 |
| Whippet | Julia | https://github.com/timbitz/Whippet.jl, accessed on 1 November 2023 | |
| Recommended APA Tools RNA-seq | TAPAS | R package | https://doi.org/10.1093/bioinformatics/bty110, accessed on 1 November 2023 |
| DaPars2 | Python | https://doi.org/10.1038/ncomms6274, accessed on 1 November 2023 | |
| APAtrap | R package/PERL | https://doi.org/10.1093/bioinformatics/bty029, accessed on 1 November 2023 | |
| QAPA | R package/Python | https://doi.org/10.1186/s13059-018-1414-4, accessed on 1 November 2023 | |
| Recommended APA Tools scRNA-seq | scAPA | R package | https://doi.org/10.1093/nar/gkz781, accessed on 1 November 2023 |
| scAPAtrap | R package | https://doi.org/10.1093/bib/bbaa273, accessed on 1 November 2023 | |
| Web Tools | eVITTA | https://doi.org/10.1093/nar/gkab366, accessed on 1 November 2023 | |
| SpliceTools | PERL for download | https://doi.org/10.1093/nar/gkad111, accessed on 1 November 2023 | |
| APAview | Jinja/Python | https://doi.org/10.3389/fgene.2022.928862, accessed on 1 November 2023 | |
| Cas-Viewer | https://doi.org/10.1186/s12920-018-0348-8, accessed on 1 November 2023 |
| Database Type | Database Name | Description | Website |
|---|---|---|---|
| AES | MutSpliceDB | Effects of mutation on splicing | https://brb.nci.nih.gov/splicing/, accessed on 1 November 2023 |
| VastDB | Splicing in multiple species | https://vastdb.crg.eu, accessed on 1 November 2023 | |
| HEXEvent | Human exon splicing | https://hexevent.mmg.uci.edu, accessed on 1 November 2023 | |
| ExonSkipDB | Exon-skipping events | https://ccsm.uth.edu/ExonSkipDB/, accessed on 1 November 2023 | |
| ClinVar | Variants with clinical phenotypes | https://www.ncbi.nlm.nih.gov/clinvar/, accessed on 1 November 2023 | |
| ATS | DBTSS | Human adult and embryonic tissues | https://dbtss.hgc.jp/, accessed on 1 November 2023 |
| refTSS | Human and mouse | https://reftss.riken.jp/, accessed on 1 November 2023 | |
| APA | PolyASite 2.0 | Sites and usage in human, mouse and worm | https://www.polyasite.unibas.ch/, accessed on 1 November 2023 |
| PolyA DB3 | Sites, cleavage, and conservation | https://exon.apps.wistar.org/PolyA_DB/, accessed on 1 November 2023 | |
| scAPAdb | Sites and usage in multiple species, single-cell data | http://www.bmibig.cn/scAPAdb/, accessed on 1 November 2023 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrion, S.A.; Michal, J.J.; Jiang, Z. Alternative Transcripts Diversify Genome Function for Phenome Relevance to Health and Diseases. Genes 2023, 14, 2051. https://doi.org/10.3390/genes14112051
Carrion SA, Michal JJ, Jiang Z. Alternative Transcripts Diversify Genome Function for Phenome Relevance to Health and Diseases. Genes. 2023; 14(11):2051. https://doi.org/10.3390/genes14112051
Chicago/Turabian StyleCarrion, Shane A., Jennifer J. Michal, and Zhihua Jiang. 2023. "Alternative Transcripts Diversify Genome Function for Phenome Relevance to Health and Diseases" Genes 14, no. 11: 2051. https://doi.org/10.3390/genes14112051