

Genome-Wide Association Study of Beta-Blocker Survival Benefit in Black and White Patients with Heart Failure with Reduced Ejection Fraction
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Discovery Dataset
2.2. Validation Dataset
2.3. Beta-Blocker Exposure
2.4. Genotyping
2.5. Statistical Analysis
3. Results
3.1. Baseline Characteristics
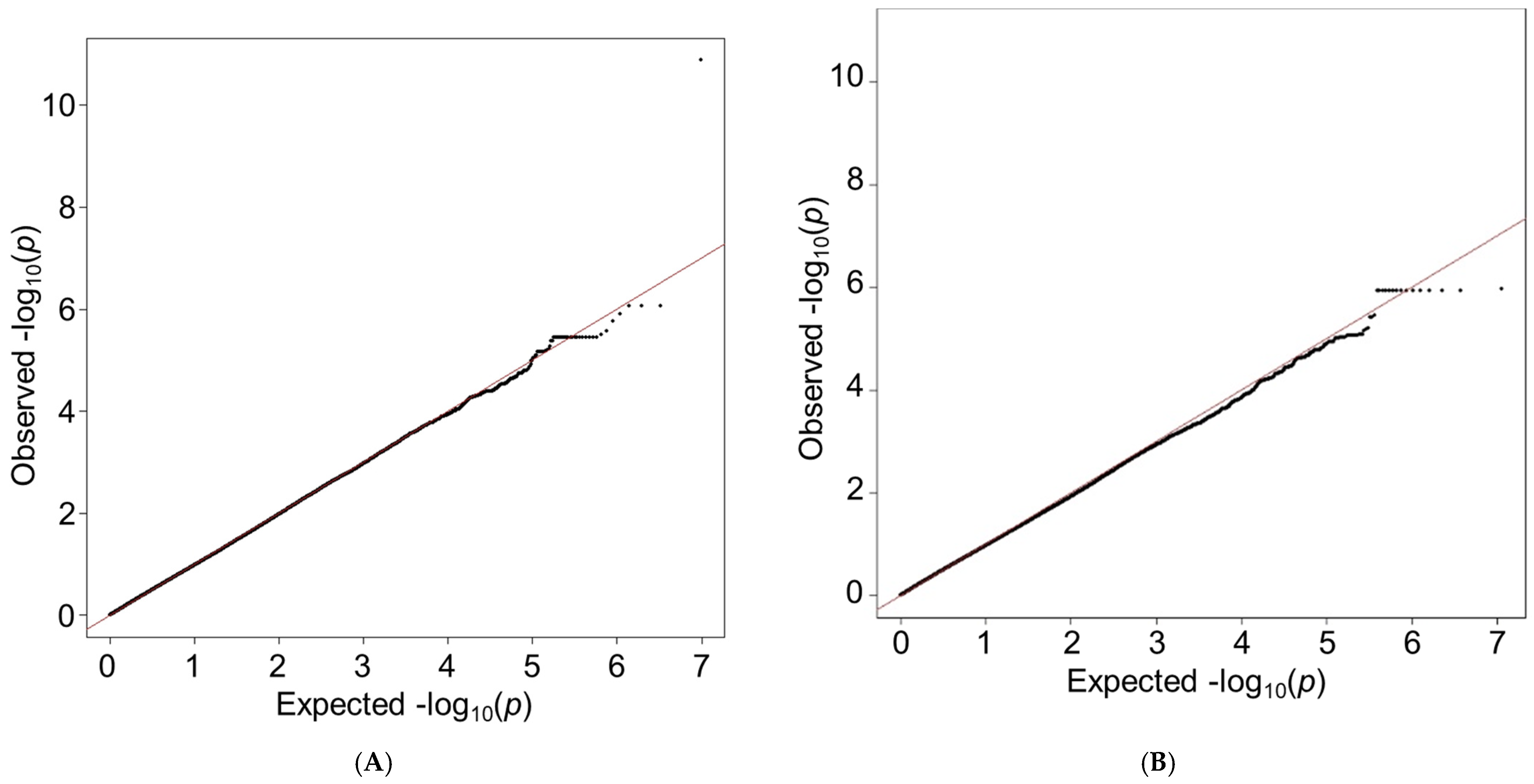
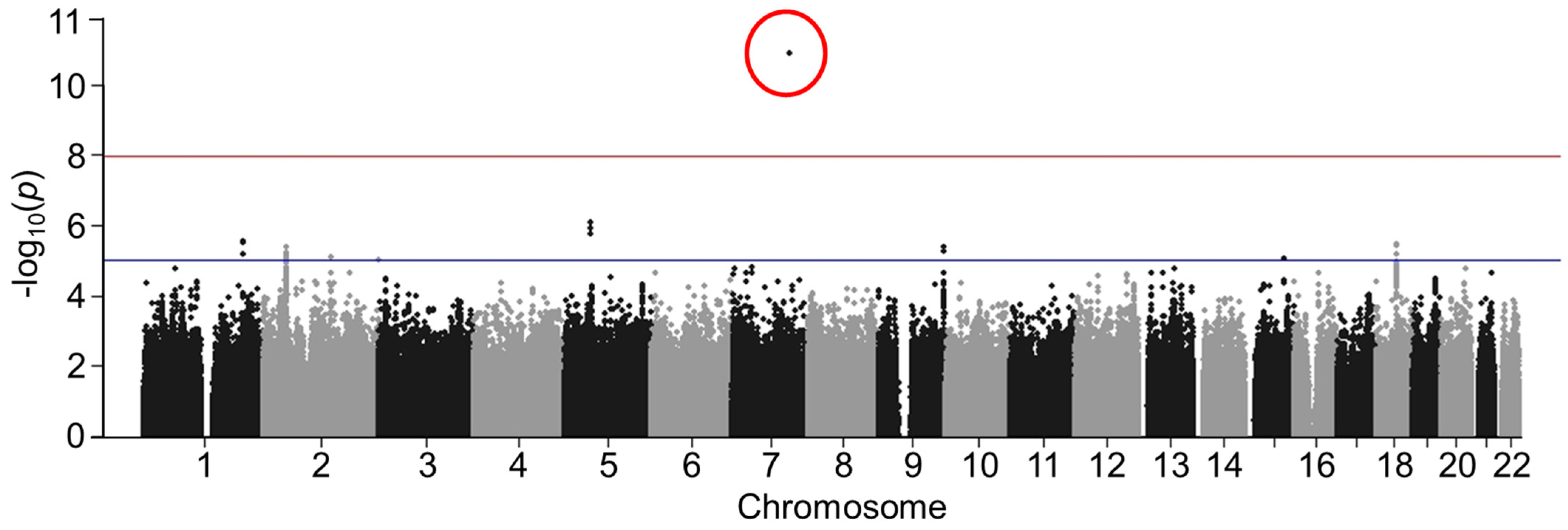
3.2. Discovery GWAS
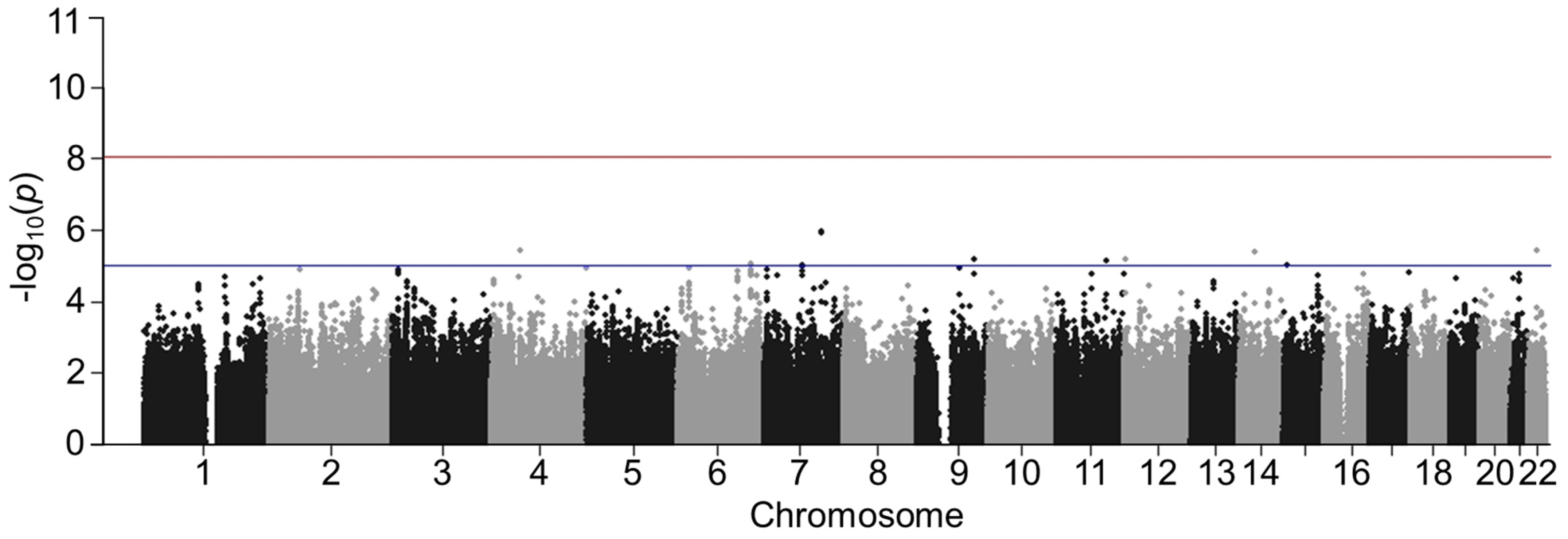
3.3. Validation of GWAS Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Savarese, G.; Lund, L.H. Global Public Health Burden of Heart Failure. Card. Fail. Rev. 2017, 3, 7–11. [Google Scholar] [CrossRef]
- Maddox, T.M.; Januzzi, J.L.; Allen, L.A.; Breathett, K.; Butler, J.; Davis, L.L.; Fonarow, G.C.; Ibrahim, N.E.; Lindenfeld, J.; Masoudi, F.A.; et al. 2021 Update to the 2017 ACC Expert Consensus Decision Pathway for Optimization of Heart Failure Treatment: Answers to 10 Pivotal Issues About Heart Failure With Reduced Ejection Fraction. J. Am. Coll. Cardiol. 2021, 77, 772–810. [Google Scholar] [CrossRef] [PubMed]
- MERIT-HF Investigators. Effect of Metoprolol CR/XL in Chronic Heart Failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 1999, 353, 2001–2007. [Google Scholar] [CrossRef]
- CIBIS Investigators and Committees. A Randomized Trial of Beta-Blockade in Heart Failure. The Cardiac Insufficiency Bisoprolol Study (CIBIS). Circulation 1994, 90, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Bristow, M.R.; Cohn, J.N.; Colucci, W.S.; Fowler, M.B.; Gilbert, E.M.; Shusterman, N.H. The Effect of Carvedilol on Morbidity and Mortality in Patients with Chronic Heart Failure. U.S. Carvedilol Heart Failure Study Group. N. Engl. J. Med. 1996, 334, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Metra, M.; Nodari, S.; Parrinello, G.; Giubbini, R.; Manca, C.; Dei Cas, L. Marked Improvement in Left Ventricular Ejection Fraction during Long-Term Beta-Blockade in Patients with Chronic Heart Failure: Clinical Correlates and Prognostic Significance. Am. Heart J. 2003, 145, 292–299. [Google Scholar] [CrossRef]
- Lupón, J.; Díez-López, C.; de Antonio, M.; Domingo, M.; Zamora, E.; Moliner, P.; González, B.; Santesmases, J.; Troya, M.I.; Bayés-Genís, A. Recovered Heart Failure with Reduced Ejection Fraction and Outcomes: A Prospective Study. Eur. J. Heart Fail. 2017, 19, 1615–1623. [Google Scholar] [CrossRef]
- Metra, M.; Giubbini, R.; Nodari, S.; Boldi, E.; Modena, M.G.; Dei Cas, L. Differential Effects of Beta-Blockers in Patients with Heart Failure: A Prospective, Randomized, Double-Blind Comparison of the Long-Term Effects of Metoprolol versus Carvedilol. Circulation 2000, 102, 546–551. [Google Scholar] [CrossRef]
- Luzum, J.A.; Peterson, E.; Li, J.; She, R.; Gui, H.; Liu, B.; Spertus, J.A.; Pinto, Y.M.; Williams, L.K.; Sabbah, H.N.; et al. Race and Beta-Blocker Survival Benefit in Patients With Heart Failure: An Investigation of Self-Reported Race and Proportion of African Genetic Ancestry. J. Am. Heart Assoc. 2018, 7, e007956. [Google Scholar] [CrossRef]
- Talameh, J.A.; Lanfear, D. Pharmacogenetics in Chronic Heart Failure: New Developments and Current Challenges. Curr. Heart Fail. Rep. 2012, 9, 23–32. [Google Scholar] [CrossRef]
- Talameh, J.A.; McLeod, H.L.; Adams, K.F.; Patterson, J.H. Genetic Tailoring of Pharmacotherapy in Heart Failure: Optimize the Old, While We Wait for Something New. J. Card. Fail. 2012, 18, 338–349. [Google Scholar] [CrossRef]
- Bristow, M.R. Beta-Adrenergic Receptor Blockade in Chronic Heart Failure. Circulation 2000, 101, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Holmer, S.R.; Hengstenberg, C.; Mayer, B.; Engel, S.; Löwel, H.; Riegger, G.A.; Schunkert, H. Marked Suppression of Renin Levels by Beta-Receptor Blocker in Patients Treated with Standard Heart Failure Therapy: A Potential Mechanism of Benefit from Beta-Blockade. J. Intern. Med. 2001, 249, 167–172. [Google Scholar] [CrossRef]
- Packer, M. The Neurohormonal Hypothesis: A Theory to Explain the Mechanism of Disease Progression in Heart Failure. J. Am. Coll. Cardiol. 1992, 20, 248–254. [Google Scholar] [CrossRef]
- Sackner-Bernstein, J.D.; Mancini, D.M. Rationale for Treatment of Patients with Chronic Heart Failure with Adrenergic Blockade. JAMA 1995, 274, 1462–1467. [Google Scholar] [CrossRef]
- McKee, P.A.; Castelli, W.P.; McNamara, P.M.; Kannel, W.B. The Natural History of Congestive Heart Failure: The Framingham Study. N. Engl. J. Med. 1971, 285, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.M.; Whellan, D.J.; Lee, K.L.; Keteyian, S.J.; Cooper, L.S.; Ellis, S.J.; Leifer, E.S.; Kraus, W.E.; Kitzman, D.W.; Blumenthal, J.A.; et al. Efficacy and Safety of Exercise Training in Patients with Chronic Heart Failure: HF-ACTION Randomized Controlled Trial. JAMA 2009, 301, 1439–1450. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, D.E.; Hrobowski, T.N.; Peterson, E.L.; Wells, K.E.; Swadia, T.V.; Spertus, J.A.; Williams, L.K. Association of β-Blocker Exposure with Outcomes in Heart Failure Differs between African American and White Patients. Circ. Heart Fail. 2012, 5, 202–208. [Google Scholar] [CrossRef]
- Lanfear, D.; Peterson, E.; Wells, K.; Williams, L.K. Discharge Medication Status Compares Poorly with Claims-Based Outpatient Medication Exposure Estimates. Circ. Cardiovasc. Qual. Outcomes 2011, 4, AP234. [Google Scholar] [CrossRef]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-Generation Genotype Imputation Service and Methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef]
- Pocock, S.J.; Ariti, C.A.; McMurray, J.J.V.; Maggioni, A.; Køber, L.; Squire, I.B.; Swedberg, K.; Dobson, J.; Poppe, K.K.; Whalley, G.A.; et al. Predicting Survival in Heart Failure: A Risk Score Based on 39 372 Patients from 30 Studies. Eur. Heart J. 2013, 34, 1404–1413. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal Components Analysis Corrects for Stratification in Genome-Wide Association Studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Patterson, N.; Price, A.L.; Reich, D. Population Structure and Eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, R.B. Propensity Score Methods for Bias Reduction in the Comparison of a Treatment to a Non-Randomized Control Group. Stat. Med. 1998, 17, 2265–2281. [Google Scholar] [CrossRef]
- Devlin, B.; Roeder, K.; Wasserman, L. Genomic Control for Association Studies: A Semiparametric Test to Detect Excess-Haplotype Sharing. Biostatistics 2000, 1, 369–387. [Google Scholar] [CrossRef]
- Rich, J.D.; Burns, J.; Freed, B.H.; Maurer, M.S.; Burkhoff, D.; Shah, S.J. Meta-Analysis Global Group in Chronic (MAGGIC) Heart Failure Risk Score: Validation of a Simple Tool for the Prediction of Morbidity and Mortality in Heart Failure With Preserved Ejection Fraction. J. Am. Heart Assoc. 2018, 7, e009594. [Google Scholar] [CrossRef] [PubMed]
- HaploReg v4.1. Available online: https://pubs.broadinstitute.org/mammals/haploreg/haploreg.php (accessed on 3 March 2022).
- Pajukanta, P.; Cargill, M.; Viitanen, L.; Nuotio, I.; Kareinen, A.; Perola, M.; Terwilliger, J.D.; Kempas, E.; Daly, M.; Lilja, H.; et al. Two Loci on Chromosomes 2 and X for Premature Coronary Heart Disease Identified in Early- and Late-Settlement Populations of Finland. Am. J. Hum. Genet. 2000, 67, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Divers, J.; Palmer, N.D.; Langefeld, C.D.; Brown, W.M.; Lu, L.; Hicks, P.J.; Smith, S.C.; Xu, J.; Terry, J.G.; Register, T.C.; et al. Genome-Wide Association Study of Coronary Artery Calcified Atherosclerotic Plaque in African Americans with Type 2 Diabetes. BMC Genet. 2017, 18, 105. [Google Scholar] [CrossRef]
- Kulminski, A.M.; He, L.; Culminskaya, I.; Loika, Y.; Kernogitski, Y.; Arbeev, K.G.; Loiko, E.; Arbeeva, L.; Bagley, O.; Duan, M.; et al. Pleiotropic Associations of Allelic Variants in a 2q22 Region with Risks of Major Human Diseases and Mortality. PLoS Genet. 2016, 12, e1006314. [Google Scholar] [CrossRef]
- Lam, C.S.P.; Gamble, G.D.; Ling, L.H.; Sim, D.; Leong, K.T.G.; Yeo, P.S.D.; Ong, H.Y.; Jaufeerally, F.; Ng, T.P.; Cameron, V.A.; et al. Mortality Associated with Heart Failure with Preserved vs. Reduced Ejection Fraction in a Prospective International Multi-Ethnic Cohort Study. Eur. Heart J. 2018, 39, 1770–1780. [Google Scholar] [CrossRef]
- Loop, M.S.; van Dyke, M.K.; Chen, L.; Safford, M.M.; Kilgore, M.L.; Brown, T.M.; Durant, R.W.; Levitan, E.B. Low Utilization of Beta-Blockers Among Medicare Beneficiaries Hospitalized for Heart Failure With Reduced Ejection Fraction. J. Card. Fail. 2019, 25, 343–351. [Google Scholar] [CrossRef]
- Aiken, L.S.; West, S.G. Multiple Regression: Testing and Interpreting Interactions; Sage: Newbury Park, CA, USA, 1991. [Google Scholar]
- Daly, A.K.; Donaldson, P.T.; Bhatnagar, P.; Shen, Y.; Pe’er, I.; Floratos, A.; Daly, M.J.; Goldstein, D.B.; John, S.; Nelson, M.R.; et al. HLA-B*5701 Genotype Is a Major Determinant of Drug-Induced Liver Injury Due to Flucloxacillin. Nat. Genet. 2009, 41, 816–819. [Google Scholar] [CrossRef]
- Lanfear, D.E.; Luzum, J.A.; She, R.; Gui, H.; Donahue, M.P.; O’Connor, C.M.; Adams, K.F.; Sanders-van Wijk, S.; Zeld, N.; Maeder, M.T.; et al. Polygenic Score for Beta-Blocker Survival Benefit in European Ancestry Patients with Reduced Ejection Fraction Heart Failure. Circ. Heart Fail. 2020, 13, e007012. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, D.E.; Luzum, J.A.; She, R.; Li, J.; Sabbah, H.N.; Zeld, N.; Liu, B.; Peterson, E.; Williams, L.K. Validation of a Polygenic Score for Beta-Blocker Survival Benefit in Patients With Heart Failure Using the United Kingdom Biobank. Circ. Genom. Precis. Med. 2023, 16, e003835. [Google Scholar] [CrossRef]
- Bis, J.C.; Sitlani, C.; Irvin, R.; Avery, C.L.; Smith, A.V.; Sun, F.; Evans, D.S.; Musani, S.K.; Li, X.; Trompet, S.; et al. Drug-Gene Interactions of Antihypertensive Medications and Risk of Incident Cardiovascular Disease: A Pharmacogenomics Study from the CHARGE Consortium. PLoS ONE 2015, 10, e0140496. [Google Scholar] [CrossRef]
- Magvanjav, O.; Gong, Y.; McDonough, C.W.; Chapman, A.B.; Turner, S.T.; Gums, J.G.; Bailey, K.R.; Boerwinkle, E.; Beitelshees, A.L.; Tanaka, T.; et al. Genetic Variants Associated With Uncontrolled Blood Pressure on Thiazide Diuretic/β-Blocker Combination Therapy in the PEAR (Pharmacogenomic Evaluation of Antihypertensive Responses) and INVEST (International Verapamil-SR Trandolapril Study) Trials. J. Am. Heart Assoc. 2017, 6, e006522. [Google Scholar] [CrossRef]
- Singh, S.; Warren, H.R.; Hiltunen, T.P.; McDonough, C.W.; El Rouby, N.; Salvi, E.; Wang, Z.; Garofalidou, T.; Fyhrquist, F.; Kontula, K.K.; et al. Genome-Wide Meta-Analysis of Blood Pressure Response to Β1-Blockers: Results From ICAPS (International Consortium of Antihypertensive Pharmacogenomics Studies). J. Am. Heart Assoc. 2019, 8, e013115. [Google Scholar] [CrossRef] [PubMed]
- Shahin, M.H.; Conrado, D.J.; Gonzalez, D.; Gong, Y.; Lobmeyer, M.T.; Beitelshees, A.L.; Boerwinkle, E.; Gums, J.G.; Chapman, A.; Turner, S.T.; et al. Genome-Wide Association Approach Identified Novel Genetic Predictors of Heart Rate Response to β-Blockers. J. Am. Heart Assoc. 2018, 7, e006463. [Google Scholar] [CrossRef]
- Chang, S.-W.; McDonough, C.W.; Gong, Y.; Johnson, T.A.; Tsunoda, T.; Gamazon, E.R.; Perera, M.A.; Takahashi, A.; Tanaka, T.; Kubo, M.; et al. Genome-Wide Association Study Identifies Pharmacogenomic Loci Linked with Specific Antihypertensive Drug Treatment and New-Onset Diabetes. Pharmacogenomics J. 2018, 18, 106–112. [Google Scholar] [CrossRef]
- Bhatt, A.S.; DeVore, A.D.; DeWald, T.A.; Swedberg, K.; Mentz, R.J. Achieving a Maximally Tolerated β-Blocker Dose in Heart Failure Patients: Is There Room for Improvement? J. Am. Coll. Cardiol. 2017, 69, 2542–2550. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Characteristics | Discovery Dataset: HFGR | Validation Dataset: HF-ACTION | ||||
|---|---|---|---|---|---|---|
| Black (n = 469) | White (n = 459) | 1 p | Black (n = 288) | White (n = 579) | 1 p | |
| Female, n (%) | 193 (41%) | 141 (31%) | 0.001 | 131 (45%) | 137 (22%) | <0.001 |
| Age, (years) | 65 ± 12 | 71 ± 10 | <0.001 | 54 ± 13 | 61 ± 12 | <0.001 |
| LVEF, (%) | 33 ± 11 | 36 ± 10 | <0.001 | 25 ± 8 | 25 ± 8 | 0.827 |
| Ischemic etiology, n (%) | 153 (33%) | 254 (55%) | <0.001 | 89 (31%) | 357 (62%) | <0.001 |
| COPD, n (%) | 105 (22%) | 111 (24%) | 0.569 | 24 (8.2%) | 85 (14%) | 0.019 |
| Atrial fibrillation or flutter, n (%) | 89 (19%) | 169 (37%) | <0.001 | 46 (16%) | 153 (25%) | 0.002 |
| Stroke, n (%) | 51 (11%) | 36 (7.8%) | 0.141 | 47 (16%) | 48 (7.8%) | <0.001 |
| Diabetes, n (%) | 219 (47%) | 172 (37%) | 0.005 | 99 (34%) | 185 (30%) | 0.288 |
| Body mass index, (kg/m2) | 31 ± 7 | 31 ± 7 | 0.140 | 33 ± 8 | 30 ± 6 | <0.001 |
| SBP, (mmHg) | 132 ± 23 | 126 ± 22 | <0.001 | 115 ± 18 | 115 ± 18 | 0.859 |
| HR, (bpm) | 72 ± 13 | 70 ± 13 | 0.011 | 71 ± 12 | 71 ± 11 | 0.688 |
| NT pro-BNP, (pg/mL) | 2962 ± 3260 | 3170 ± 3068 | 0.321 | 1266 ± 2009 | 1831 ± 2484 | 0.002 |
| Serum creatinine, (mg/dL) | 1.39 ± 1.08 | 1.18 ± 0.61 | <0.001 | 1.31 ± 0.66 | 1.33 ± 0.76 | 0.675 |
| 2 MAGGIC risk score | 17.6 ± 7.4 | 18.6 ± 7.0 | 0.028 | 18.4 ± 5.7 | 20.7 ± 6.6 | <0.001 |
| Beta-blocker exposure, (mg/day) | 0.22 ± 0.32 | 0.22 ± 0.32 | 0.911 | 0.76 ± 0.47 | 0.60 ± 0.42 | <0.001 |
| Length of follow-up, (days) | 1105 ± 695 | 1118 ± 707 | 0.780 | 984 ± 374 | 989 ± 378 | 0.853 |
| Deaths, n (%) | 112 (24%) | 116 (25%) | 0.677 | 43 (15%) | 86 (15%) | >0.999 |
| HFGR (n = 469) | HF-ACTION (n = 288) | |||||||
|---|---|---|---|---|---|---|---|---|
| Variable | Coeff. | HR | 95% CI | p-Value | Coeff. | HR | 95% CI | p-Value |
| rs16844448 × beta-blocker exposure interaction | 4.3 | 73.7 | 15.4–353.5 | 8.1 × 10−6 | 4.0 | 55.1 | 3.4–865.8 | 0.005 |
| MAGGIC (excluding beta-blocker) | 0.1 | 1.1 | 1–1.2 | <0.001 | 0.1 | 1.1 | 1–1.2 | <0.001 |
| Beta-blocker propensity score | 0.05 | 1.0 | 0.9–1.2 | 0.600 | 0.04 | 1.0 | 0.8–1.4 | 0.800 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luzum, J.A.; Campos-Staffico, A.M.; Li, J.; She, R.; Gui, H.; Peterson, E.L.; Liu, B.; Sabbah, H.N.; Donahue, M.P.; Kraus, W.E.; et al. Genome-Wide Association Study of Beta-Blocker Survival Benefit in Black and White Patients with Heart Failure with Reduced Ejection Fraction. Genes 2023, 14, 2019. https://doi.org/10.3390/genes14112019
Luzum JA, Campos-Staffico AM, Li J, She R, Gui H, Peterson EL, Liu B, Sabbah HN, Donahue MP, Kraus WE, et al. Genome-Wide Association Study of Beta-Blocker Survival Benefit in Black and White Patients with Heart Failure with Reduced Ejection Fraction. Genes. 2023; 14(11):2019. https://doi.org/10.3390/genes14112019
Chicago/Turabian StyleLuzum, Jasmine A., Alessandra M. Campos-Staffico, Jia Li, Ruicong She, Hongsheng Gui, Edward L. Peterson, Bin Liu, Hani N. Sabbah, Mark P. Donahue, William E. Kraus, and et al. 2023. "Genome-Wide Association Study of Beta-Blocker Survival Benefit in Black and White Patients with Heart Failure with Reduced Ejection Fraction" Genes 14, no. 11: 2019. https://doi.org/10.3390/genes14112019