
The Novel Link between Gene Expression Profiles of Adult T-Cell Leukemia/Lymphoma Patients’ Peripheral Blood Lymphocytes and Ferroptosis Susceptibility

Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. Sample Detection and Differential Expression Analysis
2.3. Enrichment Analysis
2.4. Protein–Protein Interaction Network Analysis and the Hub Genes
2.5. The Hub Genes and Their Interactions
2.6. Potential Pharmacological Targets
3. Results
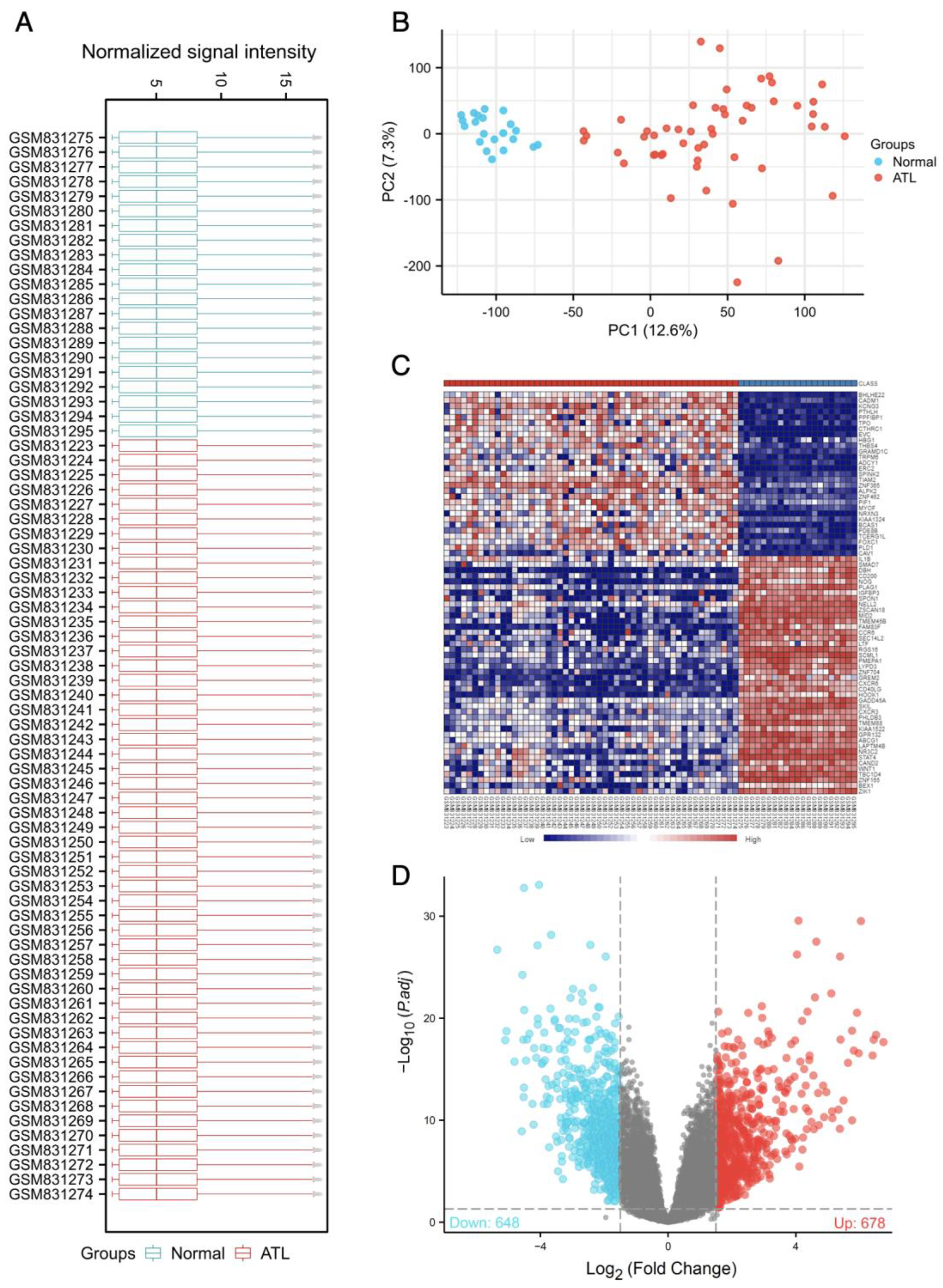
3.1. Identification of Differentially Expressed Genes of GSE33615
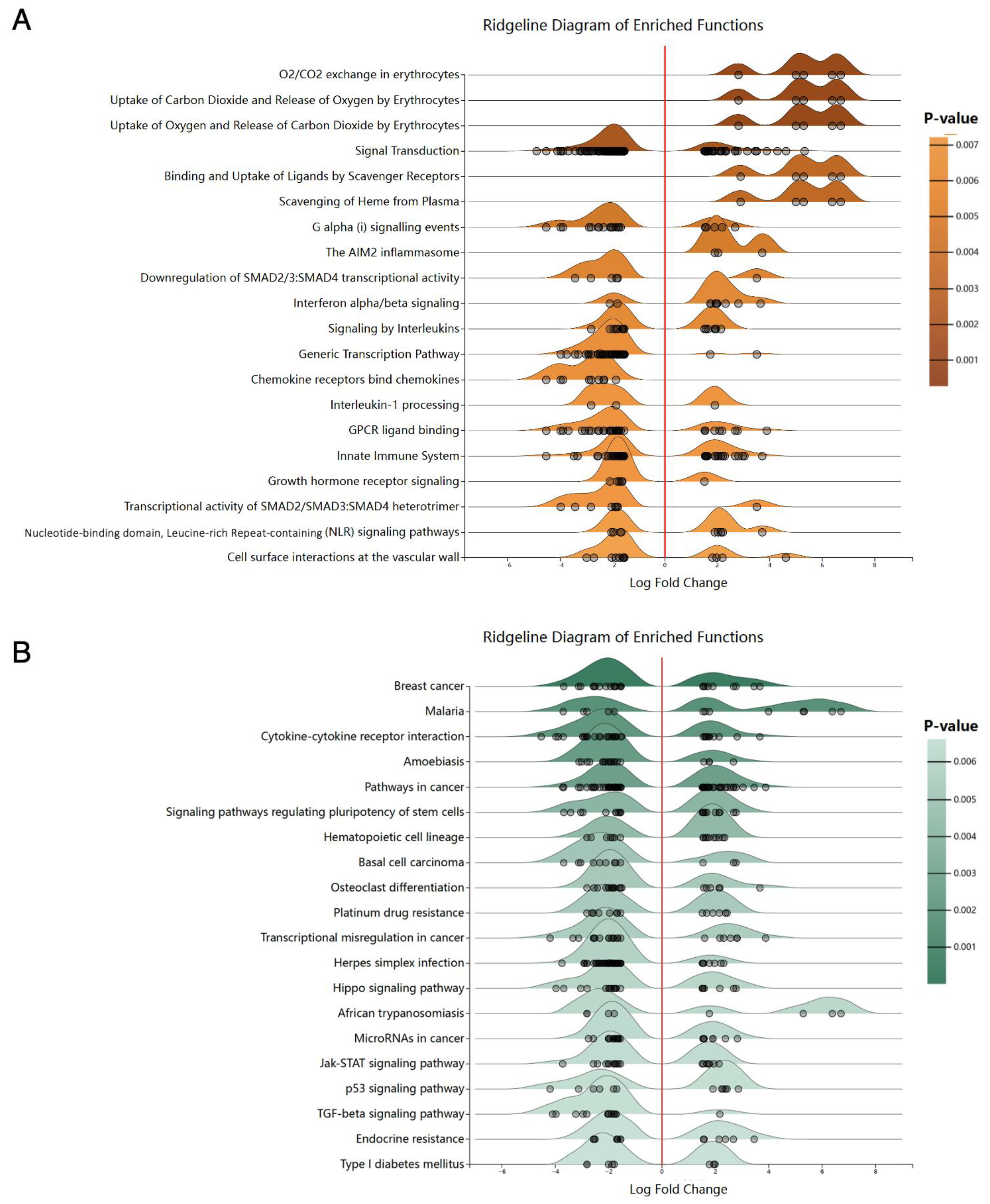
3.2. The GSEA Analysis of Differentially Expressed Genes of GSE33615
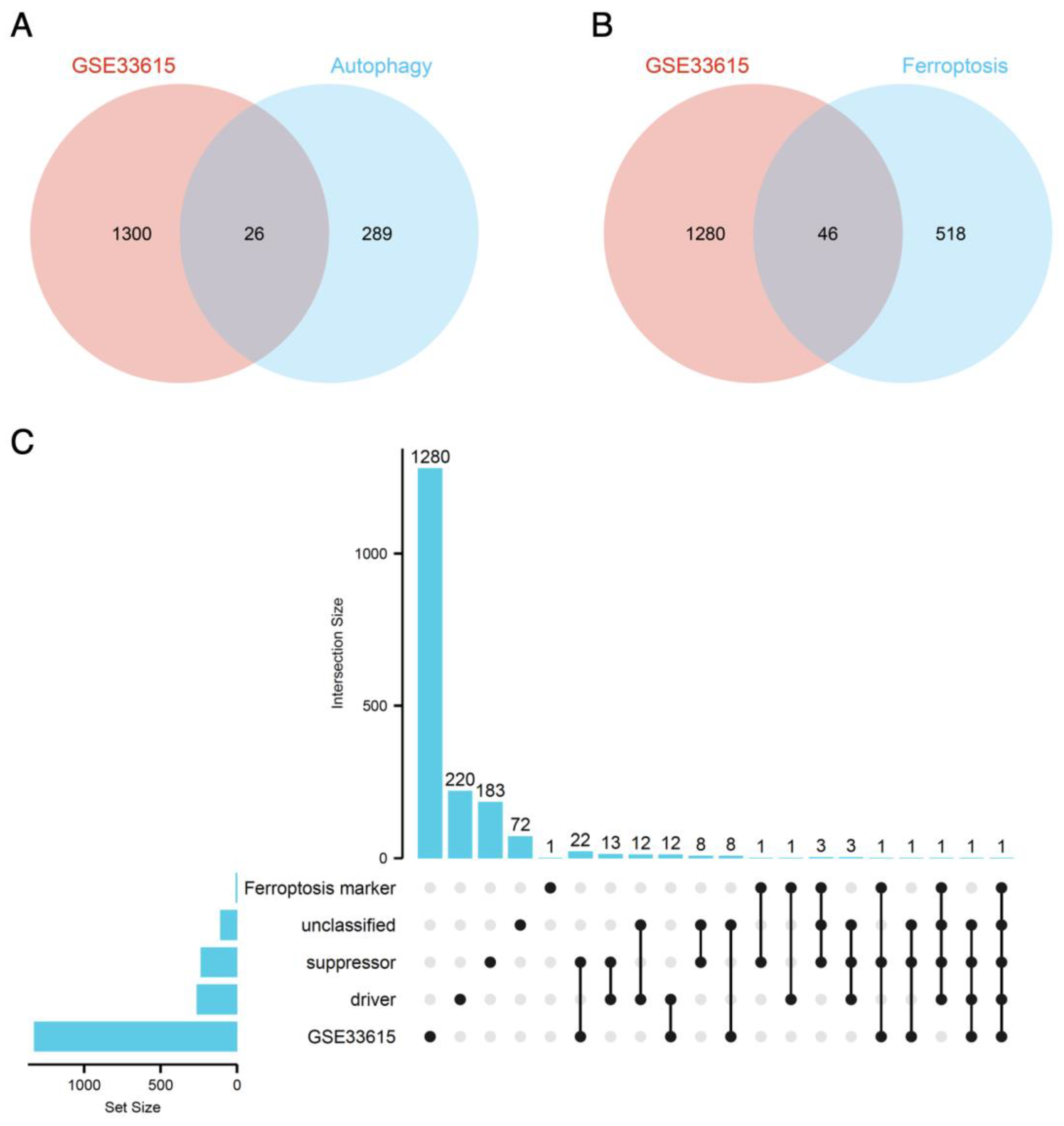
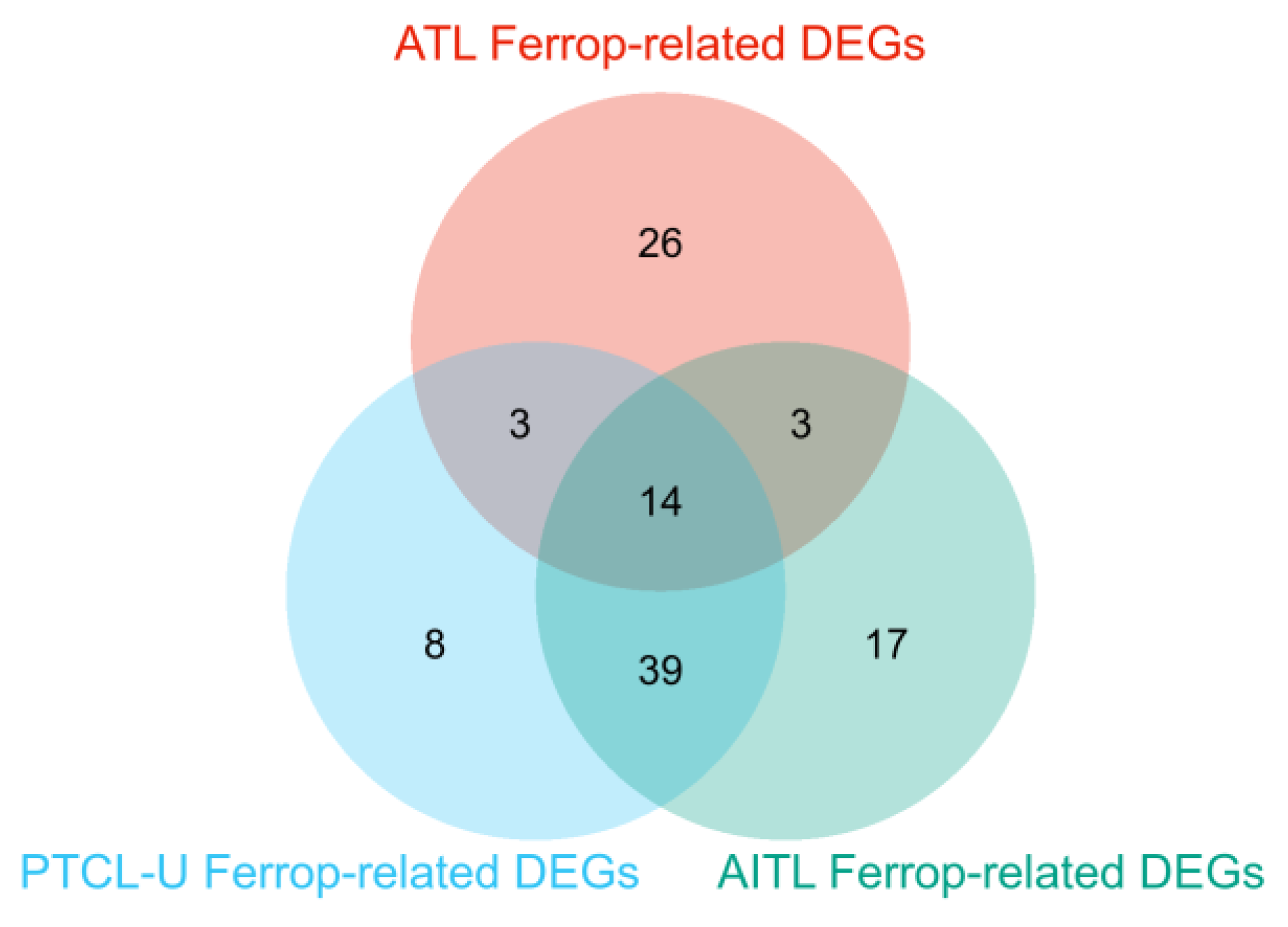
3.3. Ferroptosis-Related DEGs and Autophagy-Related DEGs
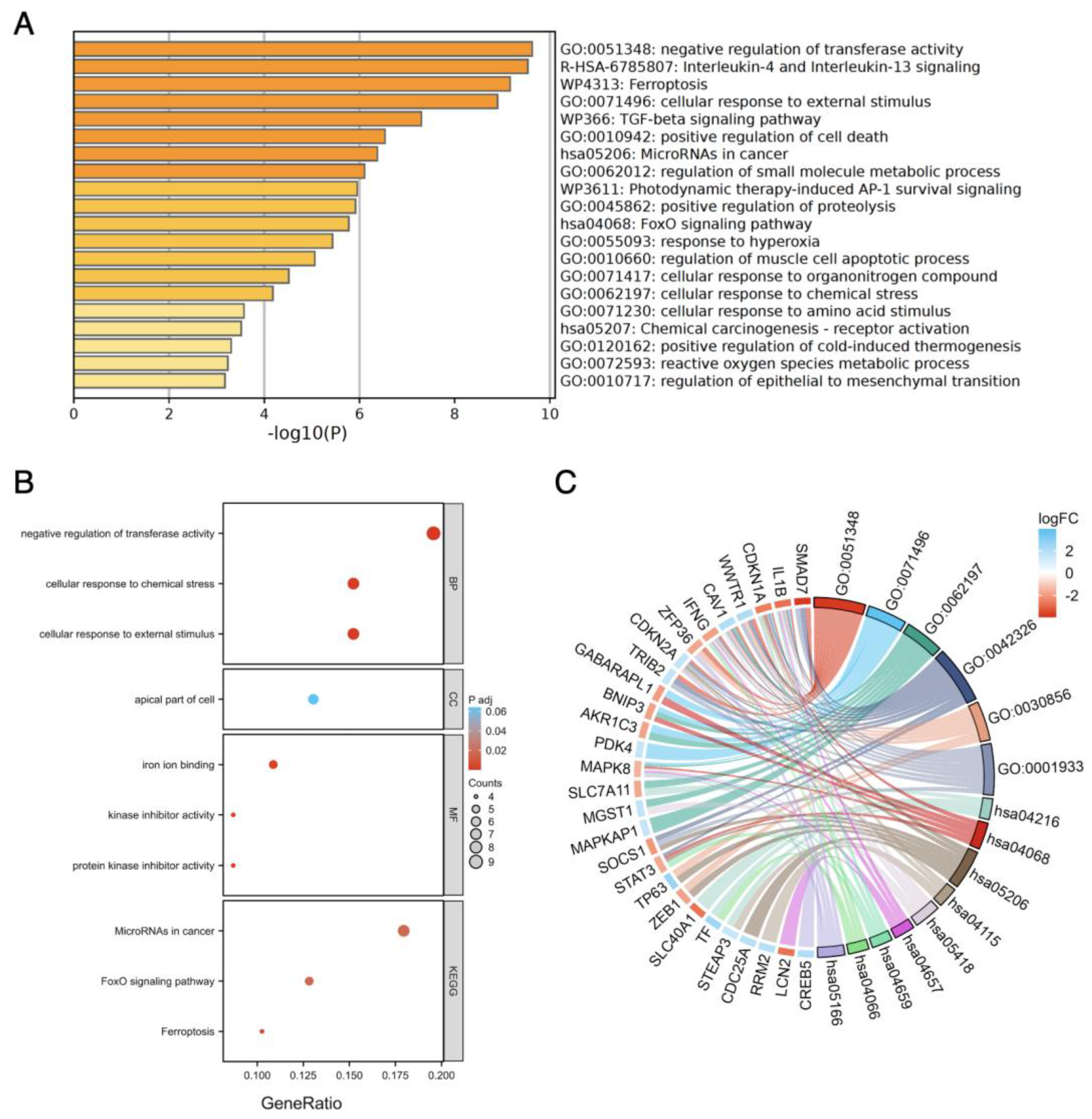
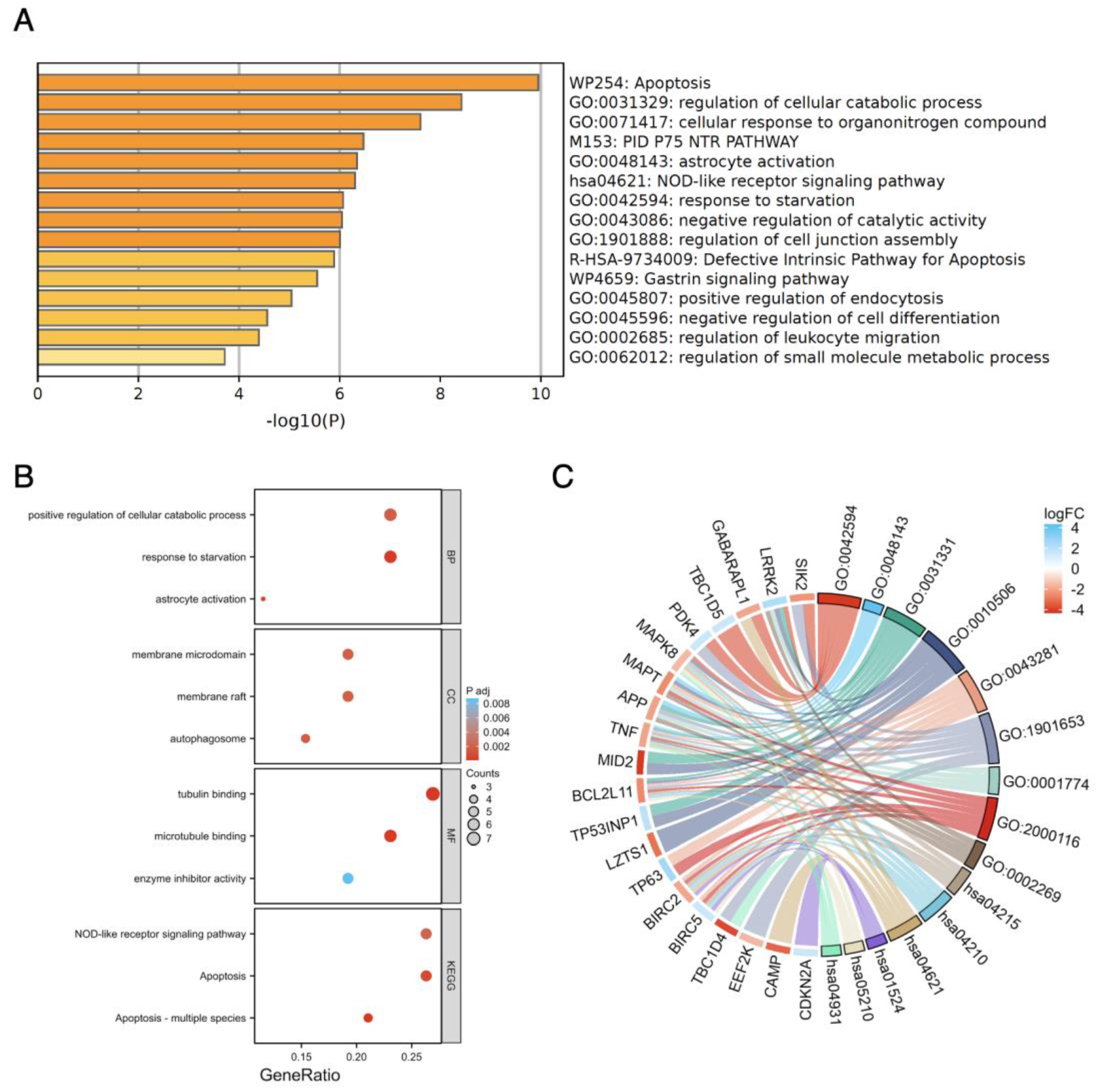
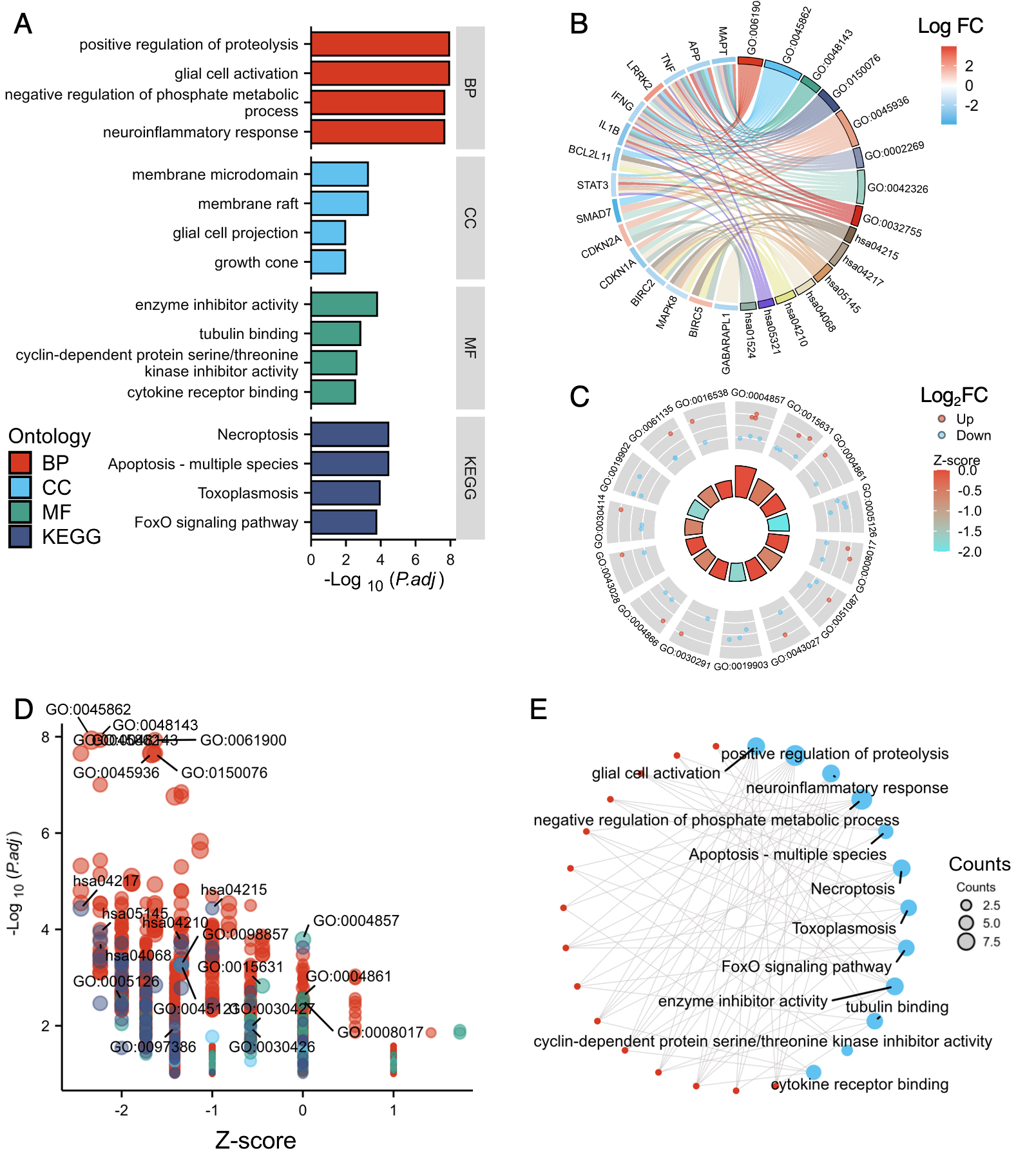
3.4. Functional Enrichment Analysis
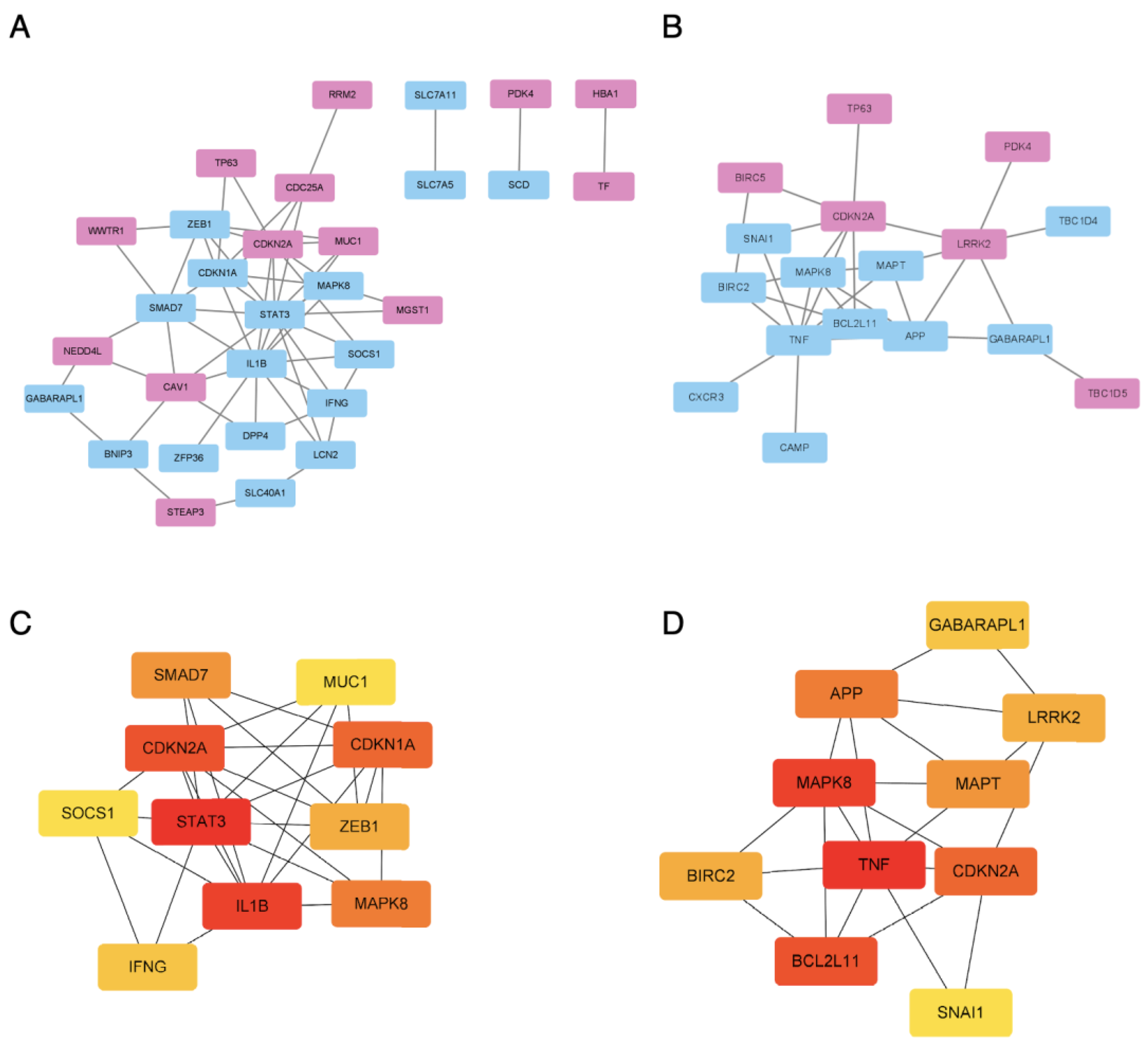
3.5. The PPI Network Analysis and Hub Gene Detection
3.6. Confirmation of the ATL-Specific Ferroptosis Signature
3.7. Construction of the Target DEGs–TF Network and the Target DEGs–ncRNA Network
3.8. Potential Pharmacology of Identified Targets DEGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ishitsuka, K.; Tamura, K. Human T-cell leukaemia virus type I and adult T-cell leukaemia-lymphoma. Lancet Oncol. 2014, 15, e517–e526. [Google Scholar] [CrossRef]
- Uchiyama, T.Y.J.; Sagawa, K.; Takatsuki, K.; Uchino, H.; Uchiyama, T.; Yodoi, J.; Sagawa, K.; Takatsuki, K.; Uchino, H. Adult T-cell leukemia: Clinical and hematologic features of 16 cases. Blood 1977, 50, 481–492. [Google Scholar] [CrossRef]
- Katsuya, H.; Ishitsuka, K.; Utsunomiya, A.; Hanada, S.; Eto, T.; Moriuchi, Y.; Saburi, Y.; Miyahara, M.; Sueoka, E.; Uike, N.; et al. Treatment and survival among 1594 patients with ATL. Blood 2015, 126, 2570–2577. [Google Scholar] [CrossRef]
- Takasaki, Y.; Iwanaga, M.; Imaizumi, Y.; Tawara, M.; Joh, T.; Kohno, T.; Yamada, Y.; Kamihira, S.; Ikeda, S.; Miyazaki, Y.; et al. Long-term study of indolent adult T-cell leukemia-lymphoma. Blood 2010, 115, 4337–4343. [Google Scholar] [CrossRef] [PubMed]
- Gessain, A.; Cassar, O. Epidemiological Aspects and World Distribution of HTLV-1 Infection. Front. Microbiol. 2012, 3, 388. [Google Scholar] [CrossRef] [PubMed]
- Arisawa, K.; Soda, M.; Akahoshi, M.; Fujiwara, S.; Uemura, H.; Hiyoshi, M.; Takeda, H.; Kashino, W.; Suyama, A. Human T-cell lymphotropic virus type-1 infection and risk of cancer: 15.4 year longitudinal study among atomic bomb survivors in Nagasaki, Japan. Cancer Sci. 2006, 97, 535–539. [Google Scholar] [CrossRef]
- Arisawa, K.; Sobue, T.; Yoshimi, I.; Soda, M.; Shirahama, S.; Doi, H.; Katamine, S.; Saito, H.; Urata, M. Human T-lymphotropic virus type-I infection, survival and cancer risk in southwestern Japan: A prospective cohort study. Cancer Causes Control 2003, 14, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, M.; Watanabe, T.; Utsunomiya, A.; Okayama, A.; Uchimaru, K.; Koh, K.R.; Ogata, M.; Kikuchi, H.; Sagara, Y.; Uozumi, K.; et al. Human T-cell leukemia virus type I (HTLV-1) proviral load and disease progression in asymptomatic HTLV-1 carriers: A nationwide prospective study in Japan. Blood 2010, 116, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, M.; Watanabe, T.; Yamaguchi, K. Adult T-cell leukemia: A review of epidemiological evidence. Front. Microbiol. 2012, 3, 322. [Google Scholar] [CrossRef]
- Pique, C.; Jones, K.S. Pathways of cell-cell transmission of HTLV-1. Front. Microbiol. 2012, 3, 378. [Google Scholar] [CrossRef] [PubMed]
- Percher, F.; Curis, C.; Pérès, E.; Artesi, M.; Rosewick, N.; Jeannin, P.; Gessain, A.; Gout, O.; Mahieux, R.; Ceccaldi, P.E.; et al. HTLV-1-induced leukotriene B4 secretion by T cells promotes T cell recruitment and virus propagation. Nat. Commun. 2017, 8, 15890. [Google Scholar] [CrossRef] [PubMed]
- Hieshima, K.; Nagakubo, D.; Nakayama, T.; Shirakawa, A.K.; Jin, Z.; Yoshie, O. Tax-inducible production of CC chemokine ligand 22 by human T cell leukemia virus type 1 (HTLV-1)-infected T cells promotes preferential transmission of HTLV-1 to CCR4-expressing CD4+ T cells. J. Immunol. 2008, 180, 931–939. [Google Scholar] [CrossRef] [PubMed]
- El Hajj, H.; Bazarbachi, A. Interplay between innate immunity and the viral oncoproteins Tax and HBZ in the pathogenesis and therapeutic response of HTLV-1 associated adult T cell leukemia. Front. Immunol. 2022, 13, 957535. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Kuang, F.; Liu, J.; Tang, D.; Kang, R. Oxidative Damage and Antioxidant Defense in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 586578. [Google Scholar] [CrossRef]
- Kim, S.E.; Zhang, L.; Ma, K.; Riegman, M.; Chen, F.; Ingold, I.; Conrad, M.; Turker, M.Z.; Gao, M.; Jiang, X.; et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat. Nanotechnol. 2016, 11, 977–985. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Doll, S.; Conrad, M. Iron and ferroptosis: A still ill-defined liaison. IUBMB Life 2017, 69, 423–434. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Aslkhodapasandhokmabad, H.; Libby, P.; Tuomilehto, J.; Lip, G.Y.H.; Penninger, J.M.; Richardson, D.R.; Tang, D.; Zhou, H.; Wang, S.; et al. Ferritinophagy and ferroptosis in the management of metabolic diseases. Trends Endocrinol. Metab. 2021, 32, 444–462. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Jiang, J.; Fu, N.; Chen, L. Targetting ferroptosis for blood cell-related diseases. J. Drug Target. 2022, 30, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 2020, 13, 110. [Google Scholar] [CrossRef]
- Beretta, G.L.; Zaffaroni, N. Radiotherapy-induced ferroptosis for cancer treatment. Front. Mol. Biosci. 2023, 10, 1216733. [Google Scholar] [CrossRef]
- Lee, J.; Roh, J.L. Unleashing Ferroptosis in Human Cancers: Targeting Ferroptosis Suppressor Protein 1 for Overcoming Therapy Resistance. Antioxidants 2023, 12, 1218. [Google Scholar] [CrossRef]
- Greco, G.; Schnekenburger, M.; Catanzaro, E.; Turrini, E.; Ferrini, F.; Sestili, P.; Diederich, M.; Fimognari, C. Discovery of Sulforaphane as an Inducer of Ferroptosis in U-937 Leukemia Cells: Expanding Its Anticancer Potential. Cancers 2021, 14, 76. [Google Scholar] [CrossRef]
- Wei, J.; Nai, G.Y.; Dai, Y.; Huang, X.J.; Xiong, M.Y.; Yao, X.Y.; Huang, Z.N.; Li, S.N.; Zhou, W.J.; Huang, Y.; et al. Dipetidyl peptidase-4 and transferrin receptor serve as prognostic biomarkers for acute myeloid leukemia. Ann. Transl. Med. 2021, 9, 1381. [Google Scholar] [CrossRef]
- Yusuf, R.Z.; Saez, B.; Sharda, A.; van Gastel, N.; Yu, V.W.C.; Baryawno, N.; Scadden, E.W.; Acharya, S.; Chattophadhyay, S.; Huang, C.; et al. Aldehyde dehydrogenase 3a2 protects AML cells from oxidative death and the synthetic lethality of ferroptosis inducers. Blood 2020, 136, 1303–1316. [Google Scholar] [CrossRef]
- Jin, L.; Tong, L. PAQR3 inhibits proliferation and aggravates ferroptosis in acute lymphoblastic leukemia through modulation Nrf2 stability. Immun. Inflamm. Dis. 2021, 9, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wu, W.; Chen, Q.; Zheng, Z.; Jiang, X.; Xue, Y.; Lin, D. TXNRD1: A Key Regulator Involved in the Ferroptosis of CML Cells Induced by Cysteine Depletion In Vitro. Oxid. Med. Cell. Longev. 2021, 2021, 7674565. [Google Scholar] [CrossRef]
- Pontel, L.B.; Bueno-Costa, A.; Morellato, A.E.; Carvalho Santos, J.; Roue, G.; Esteller, M. Acute lymphoblastic leukemia necessitates GSH-dependent ferroptosis defenses to overcome FSP1-epigenetic silencing. Redox Biol. 2022, 55, 102408. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, M.; Nakano, K.; Miyake, A.; Yamochi, T.; Kagami, Y.; Tsutsumi, A.; Matsuda, Y.; Sato-Otsubo, A.; Muto, S.; Utsunomiya, A.; et al. Polycomb-mediated loss of miR-31 activates NIK-dependent NF-kappaB pathway in adult T cell leukemia and other cancers. Cancer Cell 2012, 21, 121–135. [Google Scholar] [CrossRef]
- Fujikawa, D.; Nakagawa, S.; Hori, M.; Kurokawa, N.; Soejima, A.; Nakano, K.; Yamochi, T.; Nakashima, M.; Kobayashi, S.; Tanaka, Y.; et al. Polycomb-dependent epigenetic landscape in adult T-cell leukemia. Blood 2016, 127, 1790–1802. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, M.; Kubokawa, M.; Kuze, Y.; Suzuki, A.; Yokomizo, A.; Kobayashi, S.; Nakashima, M.; Makiyama, J.; Iwanaga, M.; Fukuda, T.; et al. Chronological genome and single-cell transcriptome integration characterizes the evolutionary process of adult T cell leukemia-lymphoma. Nat. Commun. 2021, 12, 4821. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Weisenburger, D.D.; Greiner, T.C.; Vose, J.M.; McKeithan, T.; Kucuk, C.; Geng, H.; Deffenbacher, K.; Smith, L.; Dybkaer, K.; et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood 2010, 115, 1026–1036. [Google Scholar] [CrossRef]
- Zhou, N.; Yuan, X.; Du, Q.; Zhang, Z.; Shi, X.; Bao, J.; Ning, Y.; Peng, L. FerrDb V2: Update of the manually curated database of ferroptosis regulators and ferroptosis-disease associations. Nucleic Acids Res. 2023, 51, D571–D582. [Google Scholar] [CrossRef]
- Wang, N.N.; Dong, J.; Zhang, L.; Ouyang, D.; Cheng, Y.; Chen, A.F.; Lu, A.P.; Cao, D.S. HAMdb: A database of human autophagy modulators with specific pathway and disease information. J. Cheminform. 2018, 10, 34. [Google Scholar] [CrossRef]
- Xia, J.; Fjell, C.D.; Mayer, M.L.; Pena, O.M.; Wishart, D.S.; Hancock, R.E. INMEX—A web-based tool for integrative meta-analysis of expression data. Nucleic Acids Res. 2013, 41, W63–W70. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Lyle, N.H.; Mayer, M.L.; Pena, O.M.; Hancock, R.E. INVEX—A web-based tool for integrative visualization of expression data. Bioinformatics 2013, 29, 3232–3234. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef]
- Xia, J.; Gill, E.E.; Hancock, R.E. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 2015, 10, 823–844. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Benner, M.J.; Hancock, R.E. NetworkAnalyst--integrative approaches for protein-protein interaction network analysis and visual exploration. Nucleic Acids Res. 2014, 42, W167–W174. [Google Scholar] [CrossRef] [PubMed]
- Wixon, J.; Kell, D. The Kyoto encyclopedia of genes and genomes—KEGG. Yeast 2000, 17, 48–55. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. miRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Deb, A.; Parida, S.; Mondal, S.; Khatua, S.; Ghosh, Z. LncRBase V.2: An updated resource for multispecies lncRNAs and ClinicLSNP hosting genetic variants in lncRNAs for cancer patients. RNA Biol. 2021, 18, 1136–1151. [Google Scholar] [CrossRef]
- Glazar, P.; Papavasileiou, P.; Rajewsky, N. circBase: A database for circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef]
- Savage, K.J. Therapies for peripheral T-cell lymphomas. Hematol. Am. Soc. Hematol. Educ. Program. 2011, 2011, 515–524. [Google Scholar] [CrossRef]
- Kataoka, K.; Nagata, Y.; Kitanaka, A.; Shiraishi, Y.; Shimamura, T.; Yasunaga, J.; Totoki, Y.; Chiba, K.; Sato-Otsubo, A.; Nagae, G.; et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat. Genet. 2015, 47, 1304–1315. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, M.; Fujikawa, D.; Watanabe, T.; Uchimaru, K. HTLV-1-Mediated Epigenetic Pathway to Adult T-Cell Leukemia-Lymphoma. Front. Microbiol. 2018, 9, 1686. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, M.; Jeang, K.T. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat. Rev. Cancer 2007, 7, 270–280. [Google Scholar] [CrossRef]
- Shimoyama, M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. A report from the Lymphoma Study Group (1984–87). Br. J. Haematol. 1991, 79, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn, T.; Stockwell, B.R. The development of the concept of ferroptosis. Free Radic. Biol. Med. 2019, 133, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Conrad, M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Lv, H.; Zhao, B.; Zhou, L.; Wang, S.; Luo, J.; Liu, J.; Shang, P. Iron and leukemia: New insights for future treatments. J. Exp. Clin. Cancer Res. 2019, 38, 406. [Google Scholar] [CrossRef]
- Weber, S.; Parmon, A.; Kurrle, N.; Schnutgen, F.; Serve, H. The Clinical Significance of Iron Overload and Iron Metabolism in Myelodysplastic Syndrome and Acute Myeloid Leukemia. Front. Immunol. 2020, 11, 627662. [Google Scholar] [CrossRef]
- Zain, J.; Kallam, A. Challenges in nodal peripheral T-cell lymphomas: From biological advances to clinical applicability. Front. Oncol. 2023, 13, 1150715. [Google Scholar] [CrossRef]
- Drieux, F.; Lemonnier, F.; Gaulard, P. How molecular advances may improve the diagnosis and management of PTCL patients. Front. Oncol. 2023, 13, 1202964. [Google Scholar] [CrossRef]
- Ma, L.; Huang, C.; Wang, X.J.; Xin, D.E.; Wang, L.S.; Zou, Q.C.; Zhang, Y.S.; Tan, M.D.; Wang, Y.M.; Zhao, T.C.; et al. Lysyl Oxidase 3 Is a Dual-Specificity Enzyme Involved in STAT3 Deacetylation and Deacetylimination Modulation. Mol. Cell 2017, 65, 296–309. [Google Scholar] [CrossRef]
- Morichika, K.; Karube, K.; Kayo, H.; Uchino, S.; Nishi, Y.; Nakachi, S.; Okamoto, S.; Morishima, S.; Ohshiro, K.; Nakazato, I.; et al. Phosphorylated STAT3 expression predicts better prognosis in smoldering type of adult T-cell leukemia/lymphoma. Cancer Sci. 2019, 110, 2982–2991. [Google Scholar] [CrossRef]
- Van Damme, J.; De Ley, M.; Opdenakker, G.; Billiau, A.; De Somer, P.; Van Beeumen, J. Homogeneous interferon-inducing 22K factor is related to endogenous pyrogen and interleukin-1. Nature 1985, 314, 266–268. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, K.; Yoshimoto, T.; Torigoe, K.; Kurimoto, M.; Matsui, K.; Hada, T.; Okamura, H.; Nakanishi, K. IL-12 synergizes with IL-18 or IL-1beta for IFN-gamma production from human T cells. Int. Immunol. 2000, 12, 151–160. [Google Scholar] [CrossRef] [PubMed]
- El Bougrini, J.; Pampin, M.; Chelbi-Alix, M.K. Arsenic enhances the apoptosis induced by interferon gamma: Key role of IRF-1. Cell. Mol. Biol. 2006, 52, 9–15. [Google Scholar]
- Kim, D.H.; Kong, J.H.; Byeun, J.Y.; Jung, C.W.; Xu, W.; Liu, X.; Kamel-Reid, S.; Kim, Y.K.; Kim, H.J.; Lipton, J.H. The IFNG (IFN-gamma) genotype predicts cytogenetic and molecular response to imatinib therapy in chronic myeloid leukemia. Clin. Cancer Res. 2010, 16, 5339–5350. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, V.; Gudmundsdottir, K.; Luong, P.; Leung, K.Y.; Knebel, A.; Basu, S. JNK phosphorylates Yes-associated protein (YAP) to regulate apoptosis. Cell Death Dis. 2010, 1, e29. [Google Scholar] [CrossRef]
- Williams, T.M.; Moolten, D.; Burlein, J.; Romano, J.; Bhaerman, R.; Godillot, A.; Mellon, M.; Rauscher, F.J., 3rd; Kant, J.A. Identification of a zinc finger protein that inhibits IL-2 gene expression. Science 1991, 254, 1791–1794. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.M.; Montoya, G.; Wu, Y.; Eddy, R.L.; Byers, M.G.; Shows, T.B. The TCF8 gene encoding a zinc finger protein (Nil-2-a) resides on human chromosome 10p11.2. Genomics 1992, 14, 194–196. [Google Scholar] [CrossRef]
- Hidaka, T.; Nakahata, S.; Hatakeyama, K.; Hamasaki, M.; Yamashita, K.; Kohno, T.; Arai, Y.; Taki, T.; Nishida, K.; Okayama, A.; et al. Down-regulation of TCF8 is involved in the leukemogenesis of adult T-cell leukemia/lymphoma. Blood 2008, 112, 383–393. [Google Scholar] [CrossRef]
- Starr, R.; Willson, T.A.; Viney, E.M.; Murray, L.J.; Rayner, J.R.; Jenkins, B.J.; Gonda, T.J.; Alexander, W.S.; Metcalf, D.; Nicola, N.A.; et al. A family of cytokine-inducible inhibitors of signalling. Nature 1997, 387, 917–921. [Google Scholar] [CrossRef]
- Lavens, D.; Ulrichts, P.; Catteeuw, D.; Gevaert, K.; Vandekerckhove, J.; Peelman, F.; Eyckerman, S.; Tavernier, J. The C-terminus of CIS defines its interaction pattern. Biochem. J. 2007, 401, 257–267. [Google Scholar] [CrossRef]
- Cooper, J.C.; Shi, M.; Chueh, F.Y.; Venkitachalam, S.; Yu, C.L. Enforced SOCS1 and SOCS3 expression attenuates Lck-mediated cellular transformation. Int. J. Oncol. 2010, 36, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Liu, Z.; Chen, Y. Regulation of TGF-beta signaling by Smad7. Acta Biochim. Biophys. Sin. (Shanghai) 2009, 41, 263–272. [Google Scholar] [CrossRef]
- Shehata, M.M.; Sallam, A.M.; Naguib, M.G.; El-Mesallamy, H.O. Overexpression of BAMBI and SMAD7 impacts prognosis of acute myeloid leukemia patients: A potential TERT non-canonical role. Cancer Biomark. 2021, 31, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Koyanagi, A.; Kotani, H.; Iida, Y.; Tanino, R.; Kartika, I.D.; Kishimoto, K.; Harada, M. Protective roles of cytoplasmic p21(Cip1) (/Waf1) in senolysis and ferroptosis of lung cancer cells. Cell Prolif. 2022, 55, e13326. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Tavana, O.; Chu, B.; Erber, L.; Chen, Y.; Baer, R.; Gu, W. NRF2 Is a Major Target of ARF in p53-Independent Tumor Suppression. Mol. Cell. 2017, 68, 224–232.e4. [Google Scholar] [CrossRef] [PubMed]
- Minami, J.K.; Morrow, D.; Bayley, N.A.; Fernandez, E.G.; Salinas, J.J.; Tse, C.; Zhu, H.; Su, B.; Plawat, R.; Jones, A.; et al. CDKN2A deletion remodels lipid metabolism to prime glioblastoma for ferroptosis. Cancer Cell 2023, 41, 1048–1060.e9. [Google Scholar] [CrossRef] [PubMed]
- Stroopinsky, D.; Rosenblatt, J.; Ito, K.; Mills, H.; Yin, L.; Rajabi, H.; Vasir, B.; Kufe, T.; Luptakova, K.; Arnason, J.; et al. MUC1 is a potential target for the treatment of acute myeloid leukemia stem cells. Cancer Res. 2013, 73, 5569–5579. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Komoda, M.; Yamada, Y.; Yonezawa, S.; Tsutsumida, H.; Nagai, K.; Atogami, S.; Tsuruda, K.; Osaka, A.; Sasaki, D.; et al. Aberrant overexpression of membrane-associated mucin contributes to tumor progression in adult T-cell leukemia/lymphoma cells. Leuk. Lymphoma 2011, 52, 1108–1117. [Google Scholar] [CrossRef]
- Ishida, T.; Joh, T.; Uike, N.; Yamamoto, K.; Utsunomiya, A.; Yoshida, S.; Saburi, Y.; Miyamoto, T.; Takemoto, S.; Suzushima, H.; et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: A multicenter phase II study. J. Clin. Oncol. 2012, 30, 837–842. [Google Scholar] [CrossRef]
- Tobinai, K.; Takahashi, T.; Akinaga, S. Targeting chemokine receptor CCR4 in adult T-cell leukemia-lymphoma and other T-cell lymphomas. Curr. Hematol. Malig. Rep. 2012, 7, 235–240. [Google Scholar] [CrossRef]
- Ishida, T.; Fujiwara, H.; Nosaka, K.; Taira, N.; Abe, Y.; Imaizumi, Y.; Moriuchi, Y.; Jo, T.; Ishizawa, K.; Tobinai, K.; et al. Multicenter Phase II Study of Lenalidomide in Relapsed or Recurrent Adult T-Cell Leukemia/Lymphoma: ATLL-002. J. Clin. Oncol. 2016, 34, 4086–4093. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, S.; O’Connor, O.A.; Pro, B.; Trumper, L.; Iyer, S.; Advani, R.; Bartlett, N.L.; Christensen, J.H.; Morschhauser, F.; Domingo-Domenech, E.; et al. The ECHELON-2 Trial: 5-year results of a randomized, phase III study of brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma. Ann. Oncol. 2022, 33, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Yoshimitsu, M.; Ando, K.; Ishida, T.; Yoshida, S.; Choi, I.; Hidaka, M.; Takamatsu, Y.; Gillings, M.; Lee, G.T.; Onogi, H.; et al. Oral histone deacetylase inhibitor HBI-8000 (tucidinostat) in Japanese patients with relapsed or refractory non-Hodgkin’s lymphoma: Phase I safety and efficacy. Jpn. J. Clin. Oncol. 2022, 52, 1014–1020. [Google Scholar] [CrossRef]
- Izutsu, K.; Makita, S.; Nosaka, K.; Yoshimitsu, M.; Utsunomiya, A.; Kusumoto, S.; Morishima, S.; Tsukasaki, K.; Kawamata, T.; Ono, T.; et al. An open-label, single-arm phase 2 trial of valemetostat for relapsed or refractory adult T-cell leukemia/lymphoma. Blood 2023, 141, 1159–1168. [Google Scholar]
- Yamagishi, M.; Hori, M.; Fujikawa, D.; Ohsugi, T.; Honma, D.; Adachi, N.; Katano, H.; Hishima, T.; Kobayashi, S.; Nakano, K.; et al. Targeting Excessive EZH1 and EZH2 Activities for Ab-normal Histone Methylation and Transcription Network in Malignant Lymphomas. Cell Rep. 2019, 29, 2321–2337.e7. [Google Scholar]
- Cheong, J.W.; Chong, S.Y.; Kim, J.Y.; Eom, J.I.; Jeung, H.K.; Maeng, H.Y.; Lee, S.T.; Min, Y.H. Induction of apoptosis by apicidin, a histone deacetylase inhibitor, via the activation of mitochondria-dependent caspase cascades in human Bcr-Abl-positive leukemia cells. Clin. Cancer Res. 2003, 9, 5018–5027. [Google Scholar] [PubMed]
- Laubach, J.P.; Moreau, P.; San-Miguel, J.F.; Richardson, P.G. Panobinostat for the Treatment of Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4767–4773. [Google Scholar] [CrossRef]
- Morabito, F.; Voso, M.T.; Hohaus, S.; Gentile, M.; Vigna, E.; Recchia, A.G.; Iovino, L.; Benedetti, E.; Lo-Coco, F.; Galimberti, S. Panobinostat for the treatment of acute myelogenous leukemia. Expert Opin. Investig. Drugs 2016, 25, 1117–1131. [Google Scholar] [CrossRef]
- Ikebe, E.; Kawaguchi, A.; Tezuka, K.; Taguchi, S.; Hirose, S.; Matsumoto, T.; Mitsui, T.; Senba, K.; Nishizono, A.; Hori, M.; et al. Oral administration of an HSP90 inhibitor, 17-DMAG, intervenes tumor-cell infiltration into multiple organs and improves survival period for ATL model mice. Blood Cancer J. 2013, 3, e132. [Google Scholar] [CrossRef]
- Ikebe, E.; Shimosaki, S.; Hasegawa, H.; Iha, H.; Tsukamoto, Y.; Wang, Y.; Sasaki, D.; Imaizumi, Y.; Miyazaki, Y.; Yanagihara, K.; et al. TAS-116 (pimitespib), a heat shock protein 90 inhibitor, shows efficacy in preclinical models of adult T-cell leukemia. Cancer Sci. 2022, 113, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Hasegawa, H.; Sasaki, D.; Ando, K.; Sawayama, Y.; Imanishi, D.; Taguchi, J.; Imaizumi, Y.; Hata, T.; Tsukasaki, K.; et al. Heat shock protein 90 inhibitor NVP-AUY922 exerts potent activity against adult T-cell leukemia-lymphoma cells. Cancer Sci. 2014, 105, 1601–1608. [Google Scholar] [CrossRef]
- Sharp, S.Y.; Boxall, K.; Rowlands, M.; Prodromou, C.; Roe, S.M.; Maloney, A.; Powers, M.; Clarke, P.A.; Box, G.; Sanderson, S.; et al. In vitro biological characterization of a novel, synthetic diaryl pyrazole resorcinol class of heat shock protein 90 inhibitors. Cancer Res. 2007, 67, 2206–2216. [Google Scholar] [CrossRef]
- Doi, T.; Kurokawa, Y.; Sawaki, A.; Komatsu, Y.; Ozaka, M.; Takahashi, T.; Naito, Y.; Ohkubo, S.; Nishida, T. Efficacy and safety of TAS-116, an oral inhibitor of heat shock protein 90, in patients with metastatic or unresectable gastrointestinal stromal tumour refractory to imatinib, sunitinib and regorafenib: A phase II, single-arm trial. Eur. J. Cancer 2019, 121, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Xu, G.; Cloyd, J.M.; Du, S.; Mao, Y.; Pawlik, T.M. Predicting Novel Drug Candidates for Pancreatic Neuroendocrine Tumors via Gene Signature Comparison and Connectivity Mapping. J. Gastrointest. Surg. 2022, 26, 1670–1678. [Google Scholar] [CrossRef]
- Katoh, M.; Nakagama, H. FGF receptors: Cancer biology and therapeutics. Med. Res. Rev. 2014, 34, 280–300. [Google Scholar] [CrossRef] [PubMed]
- Sampath, D.; Malik, A.; Plunkett, W.; Nowak, B.; Williams, B.; Burton, M.; Verstovsek, S.; Faderl, S.; Garcia-Manero, G.; List, A.F.; et al. Phase I clinical, pharmacokinetic, and pharmacodynamic study of the Akt-inhibitor triciribine phosphate monohydrate in patients with advanced hematologic malignancies. Leuk. Res. 2013, 37, 1461–1467. [Google Scholar] [CrossRef]
- Ranieri, R.; Pianigiani, G.; Sciabolacci, S.; Perriello, V.M.; Marra, A.; Cardinali, V.; Pierangeli, S.; Milano, F.; Gionfriddo, I.; Brunetti, L.; et al. Current status and future perspectives in targeted therapy of NPM1-mutated AML. Leukemia 2022, 36, 2351–2367. [Google Scholar] [CrossRef]
- Inchiosa, M.A., Jr. Further investigation of the potential anti-neoplastic, anti-inflammatory and immunomodulatory actions of phenoxybenzamine using the Broad Institute CLUE platform. J. Pharm. Pharmacol. Res. 2023. [Google Scholar] [CrossRef]
- Grimsby, J.; Sarabu, R.; Corbett, W.L.; Haynes, N.E.; Bizzarro, F.T.; Coffey, J.W.; Guertin, K.R.; Hilliard, D.W.; Kester, R.F.; Mahaney, P.E.; et al. Allosteric activators of glucokinase: Potential role in diabetes therapy. Science 2003, 301, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Diaz, O.; Vidalain, P.O.; Ramiere, C.; Lotteau, V.; Perrin-Cocon, L. What role for cellular metabolism in the control of hepatitis viruses? Front. Immunol. 2022, 13, 1033314. [Google Scholar] [CrossRef] [PubMed]
- Grignano, E.; Birsen, R.; Chapuis, N.; Bouscary, D. From Iron Chelation to Overload as a Therapeutic Strategy to Induce Ferroptosis in Leukemic Cells. Front. Oncol. 2020, 10, 586530. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xie, Y.; Cao, L.; Yang, L.; Yang, M.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell Oncol. 2015, 2, e1054549. [Google Scholar] [CrossRef]
- Mynott, R.L.; Habib, A.; Best, O.G.; Wallington-Gates, C.T. Ferroptosis in Haematological Malignancies and Associated Therapeutic Nanotechnologies. Int. J. Mol. Sci. 2023, 24, 7661. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
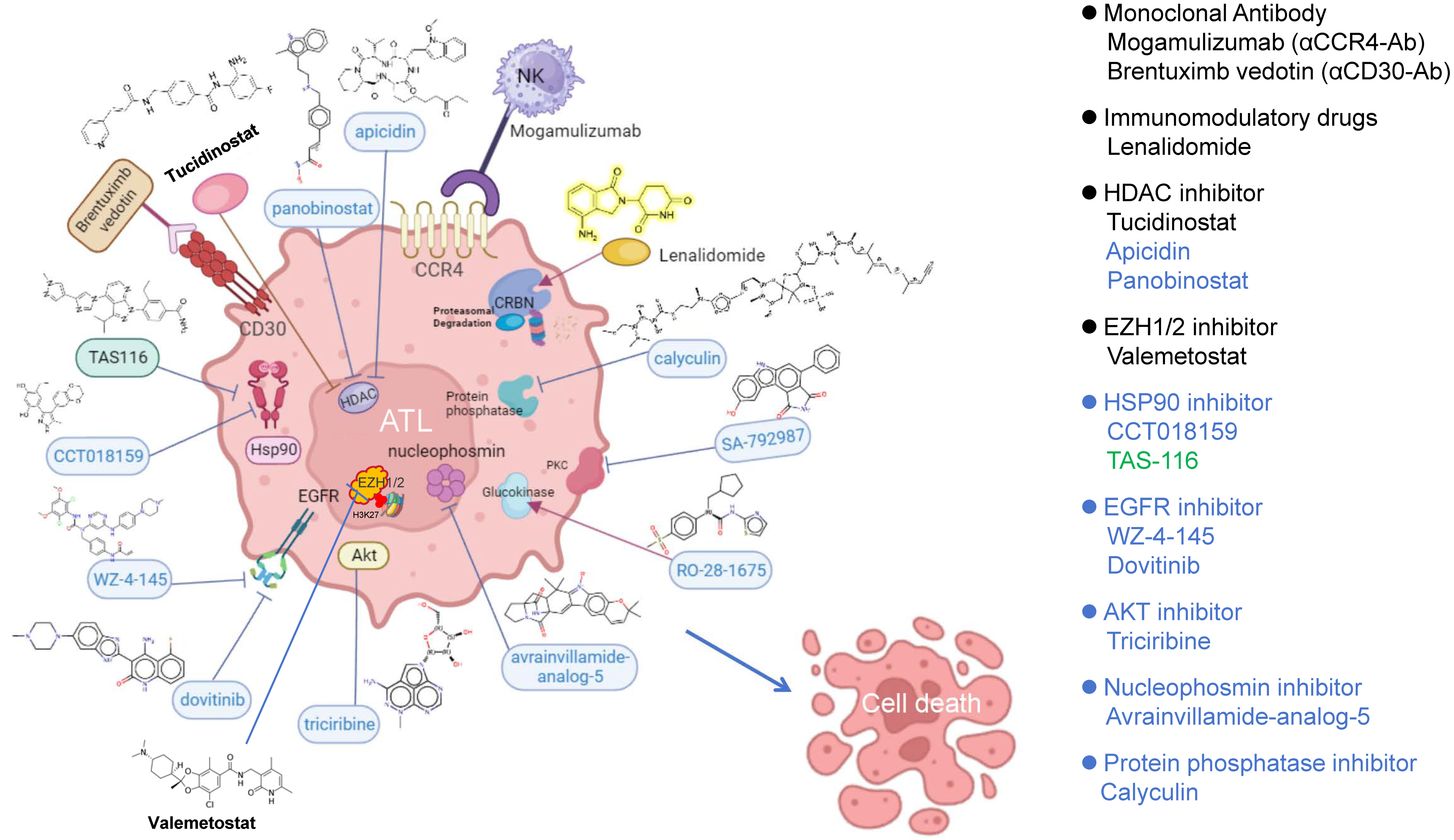{kind=link}
| Rank | Score | ID | Name | Description |
|---|---|---|---|---|
| 1 | −99.93 | BRD-A47513740 | Calyculin | Protein phosphatase inhibitor |
| 2 | −99.86 | BRD-A70731303 | Avrainvillamide-analog-5 | Nucleophosmin inhibitor |
| 3 | −99.83 | BRD-U25771771 | WZ-4-145 | EGFR inhibitor |
| 4 | −99.44 | BRD-K65503129 | CCT018159 | HSP90-inhibitor |
| 5 | −99.26 | BRD-K80431395 | Triciribine | AKT inhibitor |
| 6 | −99.15 | BRD-K85402309 | Dovitinib | EGFR inhibitor |
| 7 | −99.12 | BRD-K64606589 | Apicidin | HDAC inhibitor |
| 8 | −99.05 | BRD-K02130563 | Panobinostat | HDAC inhibitor |
| 9 | −99.03 | BRD-K82823804 | SA-792987 | PKC inhibitor |
| 10 | −99.01 | BRD-K21672174 | RO-28-1675 | Glucokinase activator |
| 11 | −98.98 | BRD-K50168500 | Canertinib | EGFR inhibitor |
| 12 | −98.94 | BRD-K68336408 | Tyrphostin-AG-1478 | EGFR inhibitor |
| 13 | −98.91 | BRD-K12502280 | TG-101348 | FLT3 inhibitor |
| 14 | −98.91 | BRD-K39120595 | Bithionol | Autotaxin inhibitor |
| 15 | −98.77 | BRD-K98490050 | Amsacrine | Topoisomerase inhibitor |
| 16 | −98.77 | BRD-K81418486 | Vorinostat | HDAC inhibitor |
| 17 | −98.72 | BRD-A17065207 | Brefeldin-a | Protein synthesis inhibitor |
| 18 | −98.70 | BRD-K10705233 | GW-405833 | Cannabinoid receptor agonist |
| 19 | −98.64 | BRD-K51575138 | TPCA-1 | IKK inhibitor |
| 20 | −98.61 | BRD-K04853698 | LDN-193189 | Serine/threonine kinase inhibitor |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Iha, H. The Novel Link between Gene Expression Profiles of Adult T-Cell Leukemia/Lymphoma Patients’ Peripheral Blood Lymphocytes and Ferroptosis Susceptibility. Genes 2023, 14, 2005. https://doi.org/10.3390/genes14112005
Wang Y, Iha H. The Novel Link between Gene Expression Profiles of Adult T-Cell Leukemia/Lymphoma Patients’ Peripheral Blood Lymphocytes and Ferroptosis Susceptibility. Genes. 2023; 14(11):2005. https://doi.org/10.3390/genes14112005
Chicago/Turabian StyleWang, Yu, and Hidekatsu Iha. 2023. "The Novel Link between Gene Expression Profiles of Adult T-Cell Leukemia/Lymphoma Patients’ Peripheral Blood Lymphocytes and Ferroptosis Susceptibility" Genes 14, no. 11: 2005. https://doi.org/10.3390/genes14112005