
Identification of the RPGR Gene Pathogenic Variants in a Cohort of Polish Male Patients with Retinitis Pigmentosa Phenotype

 , and
, and 
Abstract
:1. Introduction
2. Material and Methods
2.1. Genetic Testing
2.2. Clinical Data Collection
3. Results
3.1. Genetic Testing Results
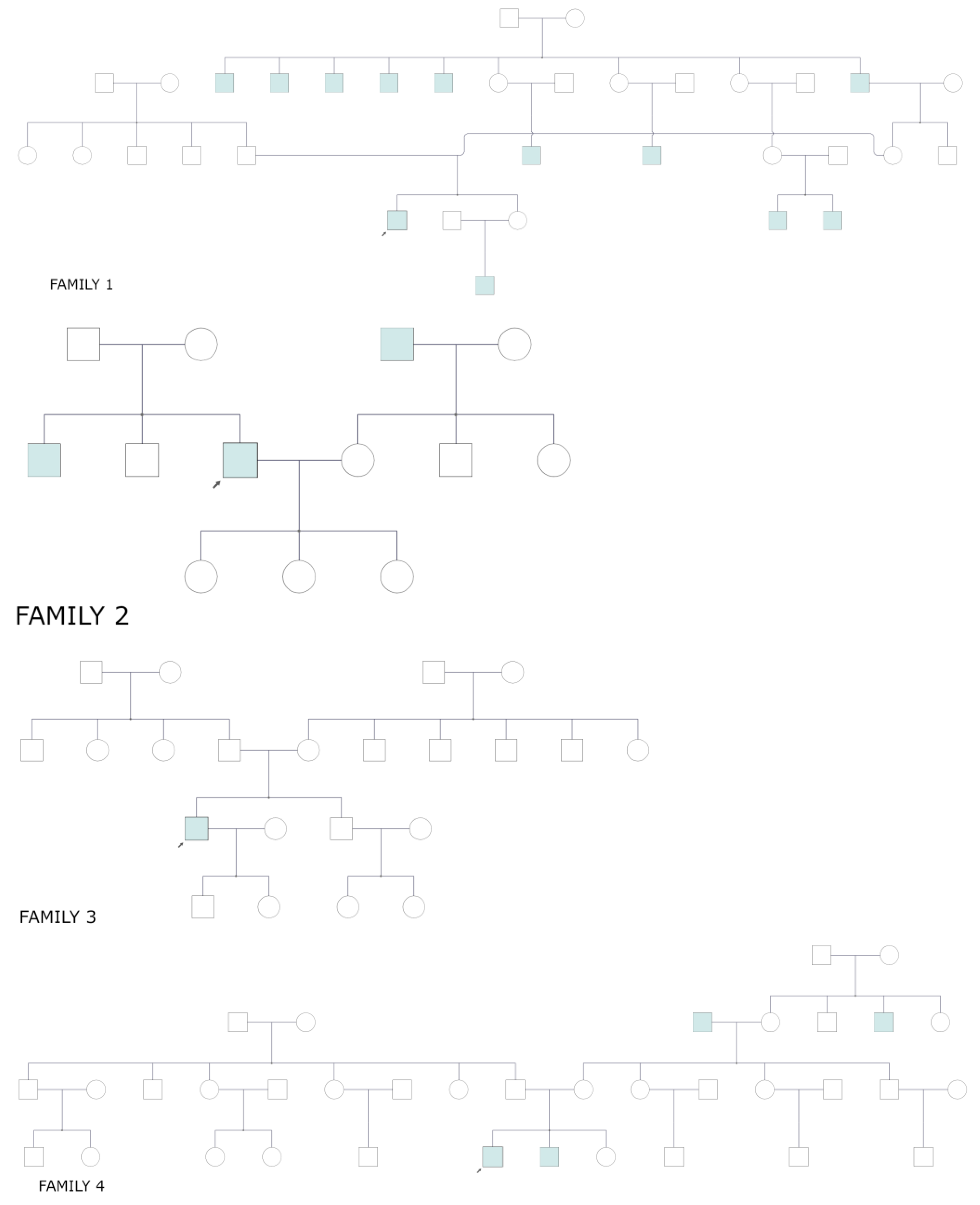
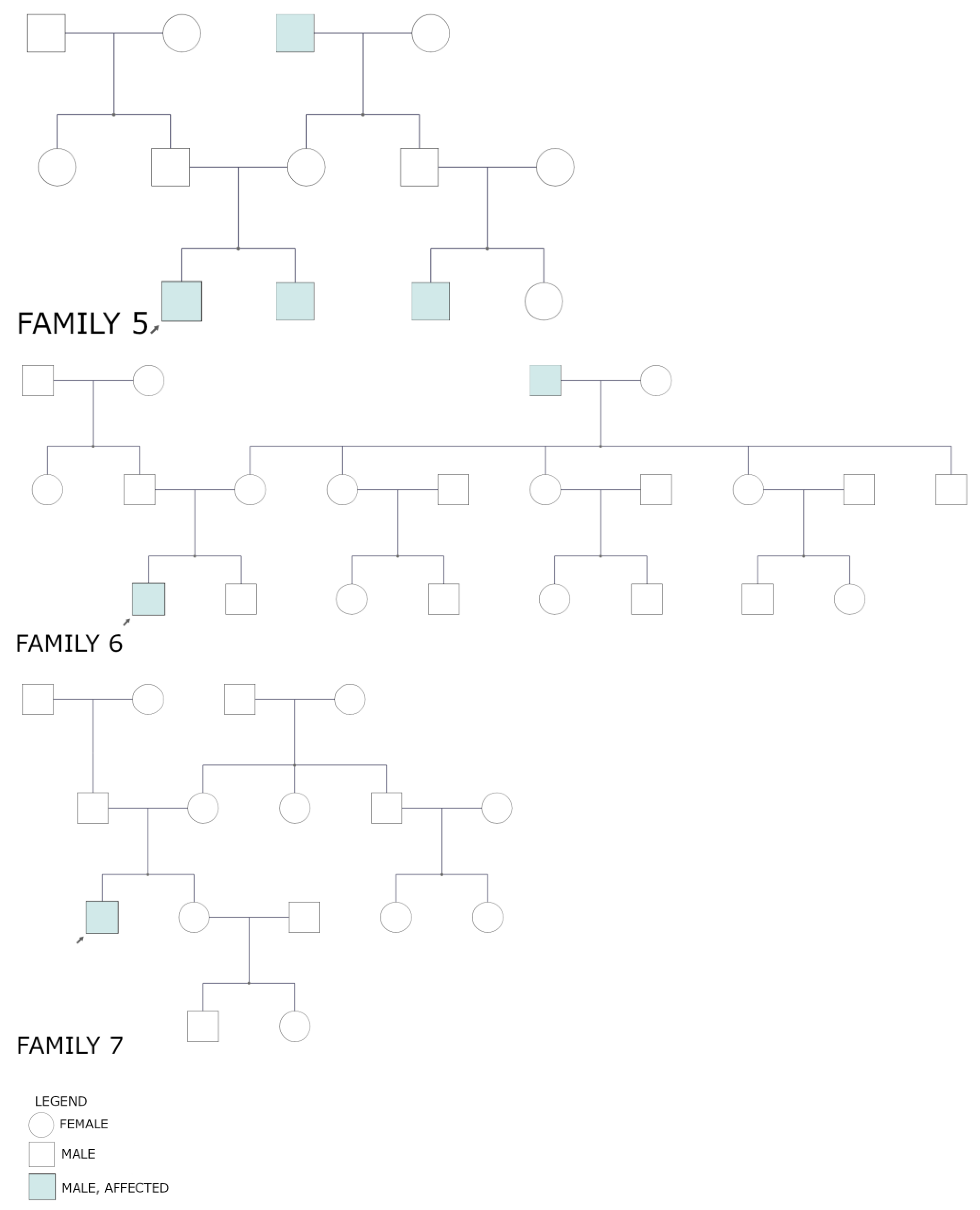
3.2. Pedigrees
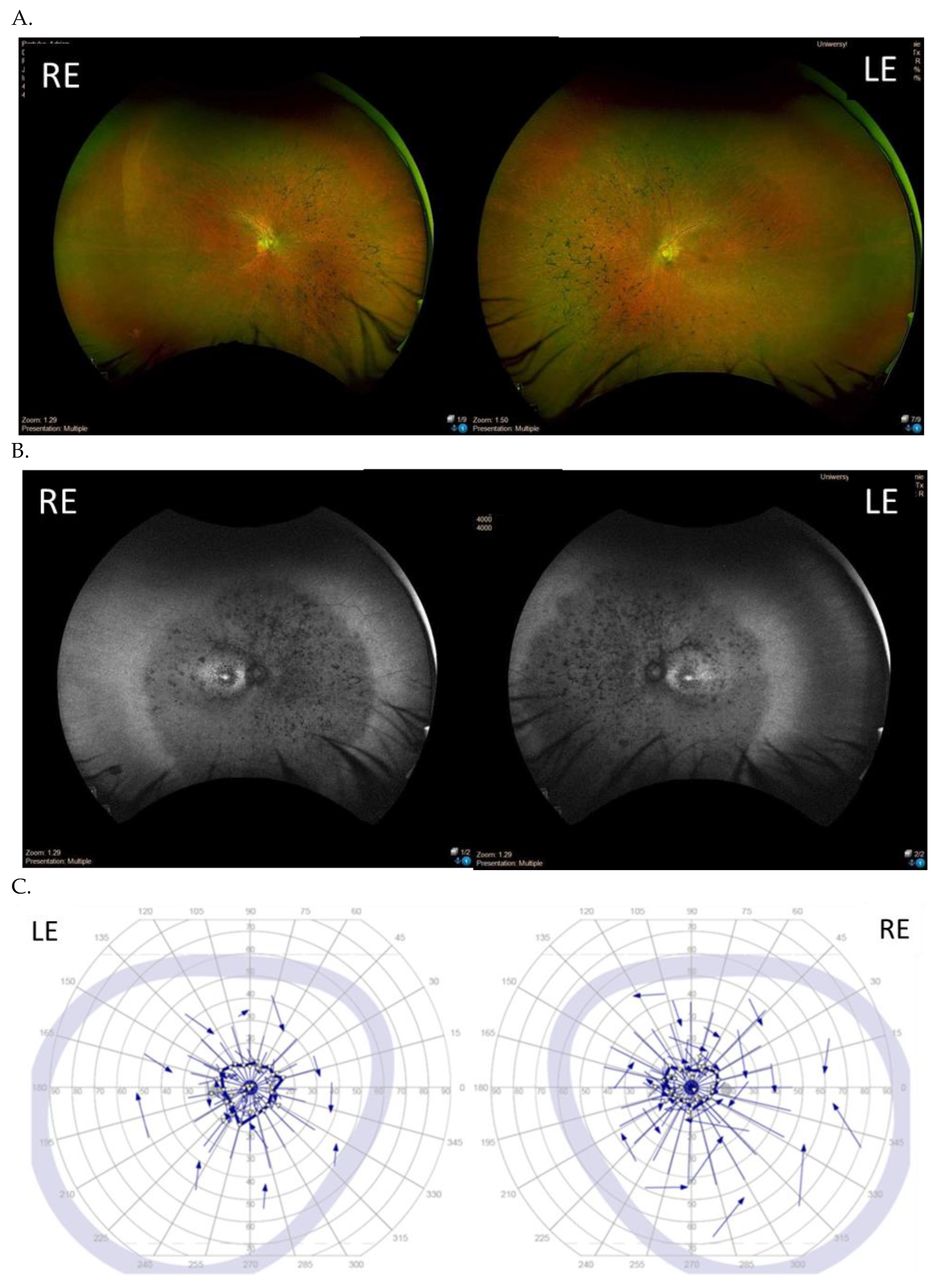
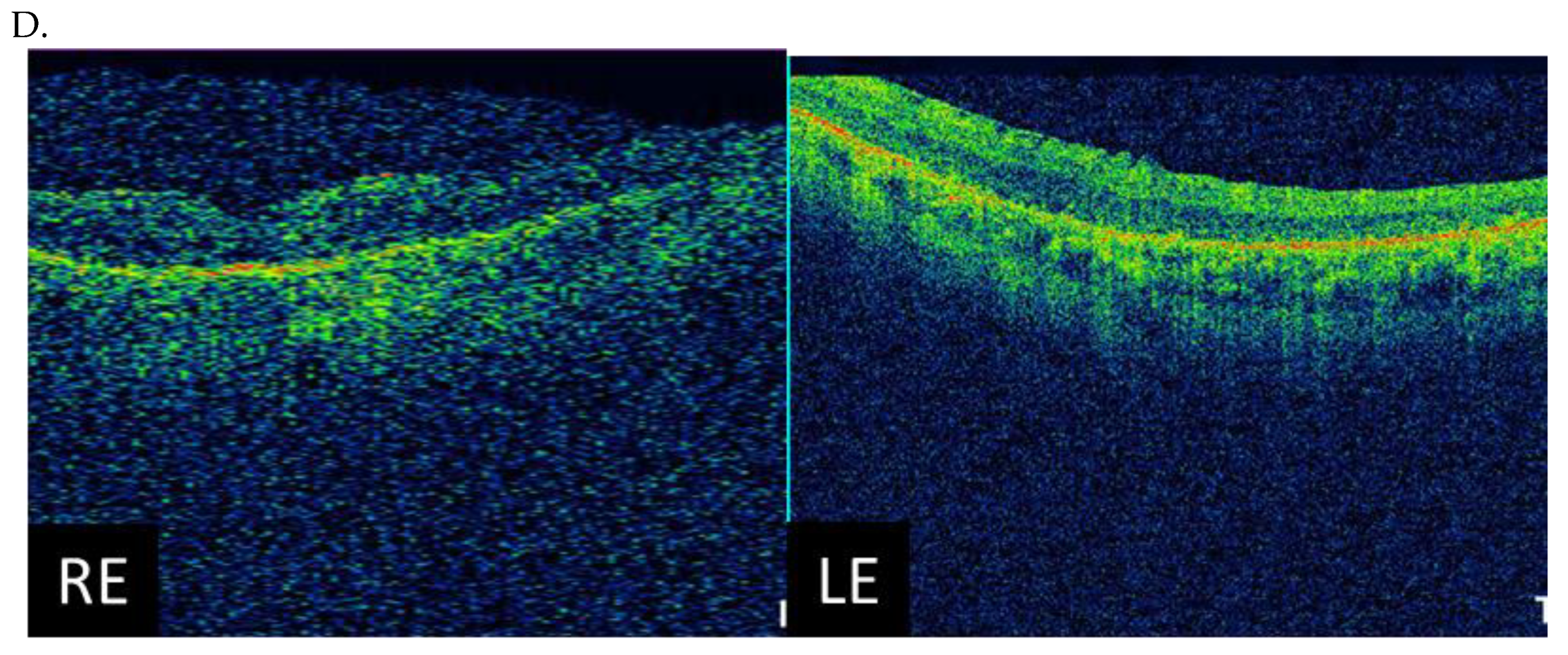
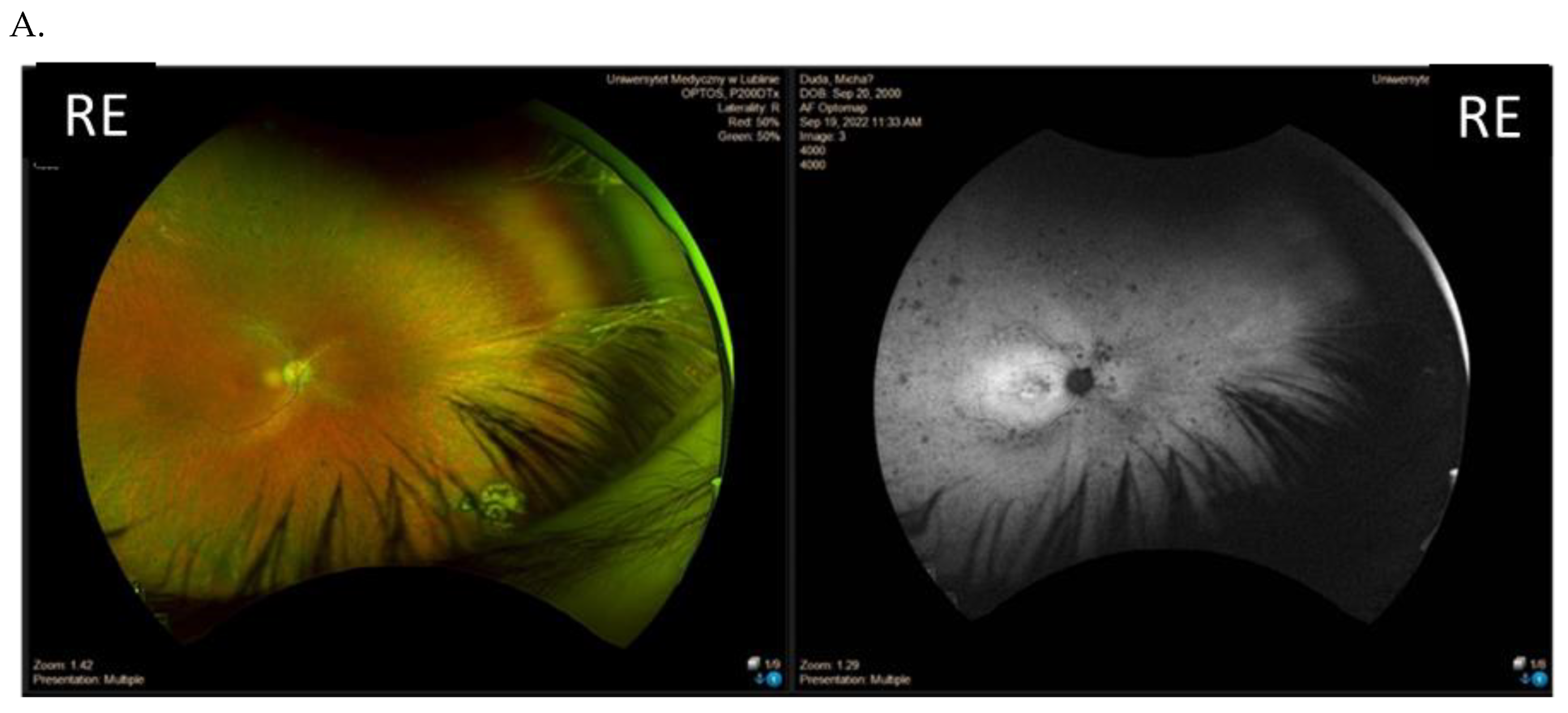
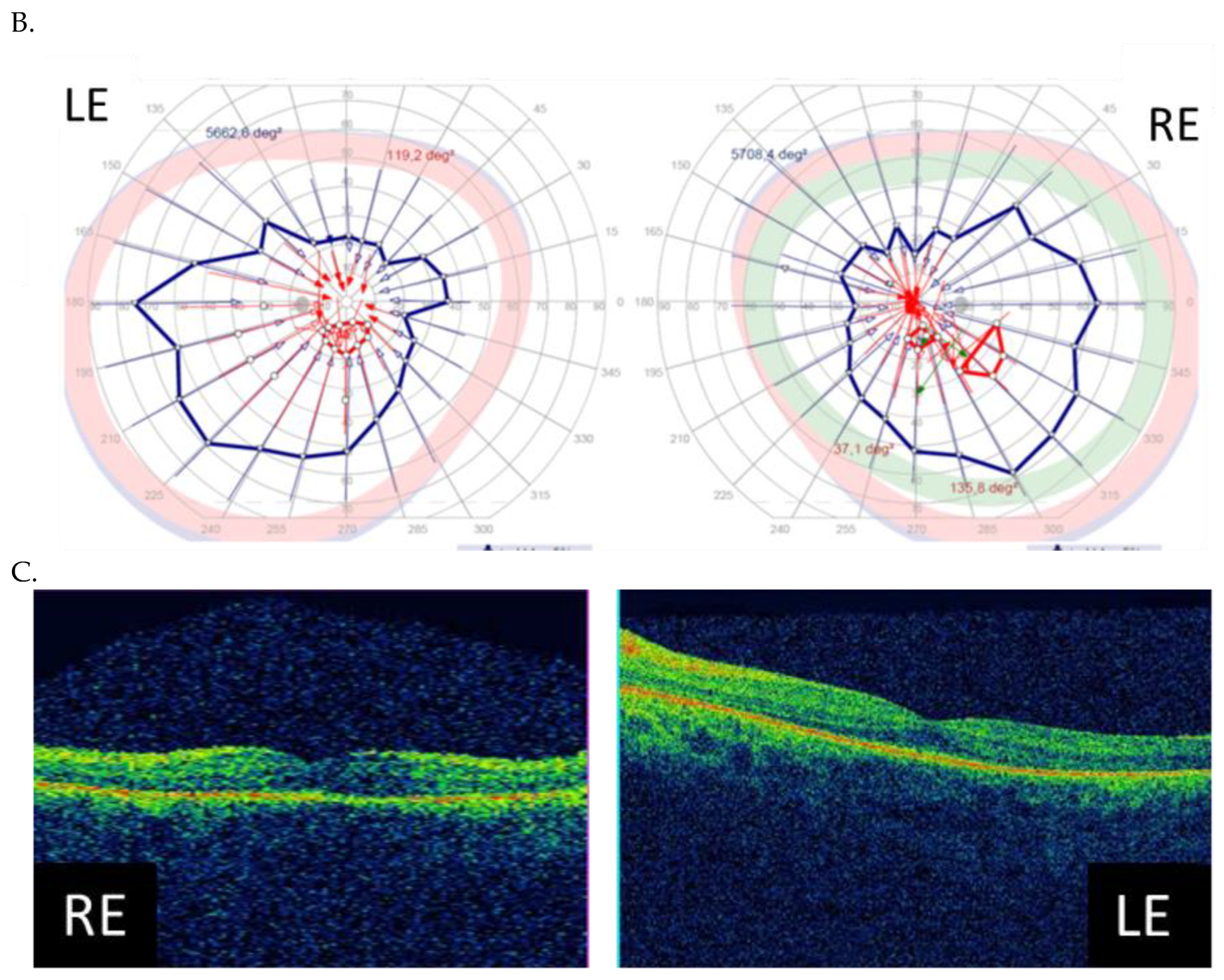
3.3. Clinical Examination Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Ben-Yosef, T.; Goldenberg-Cohen, N.; Pras, E.; Gradstein, L.; Soudry, S.; Mezer, E.; Zur, D.; Abbasi, A.H.; Zeitz, C.; et al. A nationwide genetic analysis of inherited retinal diseases in Israel as assessed by the Israeli inherited retinal disease consortium (IIRDC). Hum. Mutat. 2020, 41, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Daiger, S.P. RetNet. The Retinal Information Network. The University of Texas Health Science Center at Houston. Available online: https://web.sph.uth.edu/RetNet/ (accessed on 7 October 2022).
- Gocuk, S.A.; Jolly, J.K.; Edwards, T.L.; Ayton, L.N. Female carriers of X-linked inherited retinal diseases—Genetics, diagnosis, and potential therapies. Prog. Retin. Eye Res. 2023, 96, 101190. [Google Scholar] [CrossRef] [PubMed]
- Vervoort, R.; Lennon, A.; Bird, A.C.; Tulloch, B.; Axton, R.; Miano, M.G.; Meindl, A.; Meitinger, T.; Ciccodicola, A.; Wright, A.F. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat. Genet. 2000, 25, 462–466. [Google Scholar] [CrossRef]
- Breuer, D.K.; Yashar, B.M.; Filippova, E.; Hiriyanna, S.; Lyons, R.H.; Mears, A.J.; Asaye, B.; Acar, C.; Vervoort, R.; Wright, A.F.; et al. A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am. J. Hum. Genet. 2002, 70, 1545–1554. [Google Scholar] [CrossRef]
- Bader, I.; Brandau, O.; Achatz, H.; Apfelstedt-Sylla, E.; Hergersberg, M.; Lorenz, B.; Wissinger, B.; Wittwer, B.; Rudolph, G.; Meindl, A.; et al. X-linked retinitis pigmentosa: Rpgr mutations in most families with definite X linkage and clustering of mutations in a short sequence stretch of exon ORF15. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1458. [Google Scholar] [CrossRef]
- Branham, K.; Othman, M.; Brumm, M.; Karoukis, A.J.; Atmaca-Sonmez, P.; Yashar, B.M.; Schwartz, S.B.; Stover, N.B.; Trzupek, K.; Wheaton, D.; et al. Mutations in rpgr and rp2 account for 15% of males with simplex retinal degenerative disease. Investig. Ophthalmol. Vis. Sci. 2012, 53, 8232. [Google Scholar] [CrossRef]
- Bird, A.C. X-linked retinitis pigmentosa. Br. J. Ophthalmol. 1975, 59, 177–199. [Google Scholar] [CrossRef]
- Huang, W.C.; Wright, A.F.; Roman, A.J.; Cideciyan, A.V.; Manson, F.D.; Gewaily, D.Y.; Schwartz, S.B.; Sadigh, S.; Limberis, M.P.; Bell, P.; et al. RPGR-associated retinal degeneration in human X-linked RP and a murine model. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5594. [Google Scholar] [CrossRef]
- Neidhardt, J.; Glaus, E.; Lorenz, B.; Netzer, C.; Li, Y.; Schambeck, M.; Wittmer, M.; Feil, S.; Kirschner-Schwabe, R.; Rosenberg, T.; et al. Identification of novel mutations in X-linked retinitis pigmentosa families and implications for diagnostic testing. Mol. Vis. 2008, 14, 1081–1093. [Google Scholar]
- Hong, D.-H.; Pawlyk, B.; Sokolov, M.; Strissel, K.J.; Yang, J.; Tulloch, B.; Wright, A.F.; Arshavsky, V.Y.; Li, T. RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2413. [Google Scholar] [CrossRef] [PubMed]
- Roepman, R. The retinitis pigmentosa GTPase regulator (RPGR) interacts with novel transport-like proteins in the outer segments of Rod Photoreceptors. Hum. Mol. Gen. 2000, 9, 2095–2105. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, R.; Rosenberg, T.; Schultz-Heienbrok, R.; Lenzner, S.; Feil, S.; Roepman, R.; Cremers, F.P.; Ropers, H.-H.; Berger, W. RPGR transcription studies in mouse and human tissues reveal a retina-specific isoform that is disrupted in a patient with X-linked retinitis pigmentosa. Hum. Mol. Gen. 1999, 8, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Tracewska, A.M.; Kocyła-Karczmarewicz, B.; Rafalska, A.; Murawska, J.; Jakubaszko-Jabłónska, J.; Rydzanicz, M.; Stawiński, P.; Ciara, E.; Lipska-Ziętkiewicz, B.S.; Khan, M.I.; et al. Non-syndromic inherited retinal diseases in Poland: Genes, mutations, and phenotypes. Mol. Vis. 2021, 27, 457–465. [Google Scholar]
- Wawrocka, A.; Walczak-Sztulpa, J.; Skorczyk-Werner, A.; Kuszel, Ł.; Socha, M.; Jamsheer, A.; Krawczynski, M.R. Next-generation sequencing reveals three novel variants in Polish patients with Usher syndrome. Klin. Oczna. 2018, 2018, 189–194. [Google Scholar] [CrossRef]
- Ścieżyńska, A.; Oziębło, D.; Ambroziak, A.M.; Korwin, M.; Szulborski, K.; Krawczyński, M.; Stawiński, P.; Szaflik, J.; Szaflik, J.P.; Płoski, R.; et al. Next-generation sequencing of ABCA4: High frequency of complex alleles and novel mutations in patients with retinal dystrophies from Central Europe. Exp. Eye Res. 2016, 145, 93–99. [Google Scholar] [CrossRef]
- Skorczyk-Werner, A.; Chiang, W.-C.; Wawrocka, A.; Wicher, K.; Jarmuż-Szymczak, M.; Kostrzewska-Poczekaj, M.; Jamsheer, A.; Płoski, R.; Rydzanicz, M.; Pojda-Wilczek, D.; et al. Autosomal recessive cone-rod dystrophy can be caused by mutations in the ATF6 gene. Eur. J. Hum. Genet. 2017, 25, 1210–1216. [Google Scholar] [CrossRef]
- Churchill, J.D.; Bowne, S.J.; Sullivan, L.S.; Lewis, R.A.; Wheaton, D.K.; Birch, D.G.; Branham, K.E.; Heckenlively, J.R.; Daiger, S.P. Mutations in the X-linked retinitis pigmentosa genes rpgr and rp2 found in 8.5% of families with a provisional diagnosis of autosomal dominant retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1411. [Google Scholar] [CrossRef]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.T.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD): 2003 Update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef]
- Cehajic-Kapetanovic, J.; Martinez-Fernandez de la Camara, C.; Birtel, J.; Rehman, S.; McClements, M.E.; Charbel Issa, P.; Lotery, A.J.; MacLaren, R.E. Impaired glutamylation of RPGRORF15 underlies the cone-dominated phenotype associated with truncating distal ORF15 variants. Proc. Natl. Acad. Sci. USA 2022, 119, e2208707119. [Google Scholar] [CrossRef]
- Bellingrath, J.S.; Ochakovski, G.A.; Seitz, I.P.; Kohl, S.; Zrenner, E.; Hanig, N.; Prokisch, H.; Weber, B.H.; Downes, S.M.; Ramsden, S.; et al. High Symmetry of Visual Acuity and Visual Fields in RPGR-Linked Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2017, 58, 4457–4466. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhou, L.; Ouyang, J.; Xiao, X.; Sun, W.; Li, S.; Zhang, Q. Genotype-Phenotype Analysis of RPGR Variations: Reporting of 62 Chinese Families and a Literature Review. Front. Genet. 2021, 12, 600210. [Google Scholar] [CrossRef] [PubMed]
- Talib, M.; van Schooneveld, M.J.; Thiadens, A.A.; Fiocco, M.; Wijnholds, J.; Florijn, R.J.; Schalij-Delfos, N.E.; van Genderen, M.M.; Putter, H.; Cremers, F.P.M.; et al. Clinical and genetic characteristics of male patients with RPGR associated retinal dystrophies: A Long-Term Follow-up Study. Retina 2019, 39, 1186–1199. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Sandberg, M.A.; Rabe, V.W.; Stillberger, M.; Dryja, T.P.; Berson, E.L. RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am. J. Hum. Genet. 2003, 73, 1131–1146. [Google Scholar] [CrossRef] [PubMed]
- Wawrocka, A.; Skorczyk-Werner, A.; Wicher, K.; Niedziela, Z.; Ploski, R.; Rydzanicz, M.; Sykulski, M.; Kocieck, I.J.; Weisschuh, N.; Kohl, S.; et al. Novel variants identified with next-generation sequencing in Polish patients with cone-rod dystrophy. Mol. Vis. 2018, 24, 326–339. [Google Scholar]
- Bukowy-Bieryłło, Z.; Ziętkiewicz, E.; Loges, N.T.; Wittmer, M.; Geremek, M.; Olbrich, H.; Fliegauf, M.; Voelkel, K.; Rutkiewicz, E.; Rutland, J.; et al. RPGR mutations might cause reduced orientation of respiratory cilia. Pediatr. Pulmonol. 2013, 48, 352–363. [Google Scholar] [CrossRef]
- Ołdak, M.; Ruszkowska, E.; Siwiec, S.; Pollak, A.; Stawiński, P.; Szulborski, K.; Szaflik, J.P. ORF15 exon of the RPGR gene in retinitis pigmentosa—Technically difficult, diagnostically important. Klin. Oczn. 2016, 118, 139–143. [Google Scholar]
- Beltran, W.A.; Hammond, P.; Acland, G.M.; Aguirre, G.D. A frameshift mutation inrpgrexon orf15 causes photoreceptor degeneration and inner retina remodeling in a model of X-linked retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1669. [Google Scholar] [CrossRef]
- Zhang, Q. Different RPGR exon ORF15 mutations in canids provide insights into photoreceptor cell degeneration. Hum. Mol. Genet. 2002, 11, 993–1003. [Google Scholar] [CrossRef]
- Pelletier, V.; Jambou, M.; Delphin, N.; Zinovieva, E.; Stum, M.; Gigarel, N.; Dollfus, H.; Hamel, C.; Toutain, A.; Dufier, J.-L.; et al. Comprehensive survey of mutations in RP2 and RPGR in patients affected with distinct retinal dystrophies: Genotype–phenotype correlations and impact on genetic counseling. Hum. Mut. 2006, 28, 81–91. [Google Scholar] [CrossRef]
- Nassisi, M.; De Bartolo, G.; Mohand-Said, S.; Condroyer, C.; Antonio, A.; Lancelot, M.-E.; Bujakowska, K.; Smirnov, V.; Pugliese, T.; Neidhardt, J.; et al. Retrospective Natural History Study of RPGR-related cone- and cone-rod dystrophies while expanding the mutation spectrum of the disease. Int. J. Mol. Sci. 2022, 23, 7189. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Di Iorio, E.; Barbaro, V.; Ponzin, D.; Sorrentino, F.S.; Parmeggiani, F. Retinitis pigmentosa: Genes and disease mechanisms. Curr. Genom. 2011, 12, 238–249. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
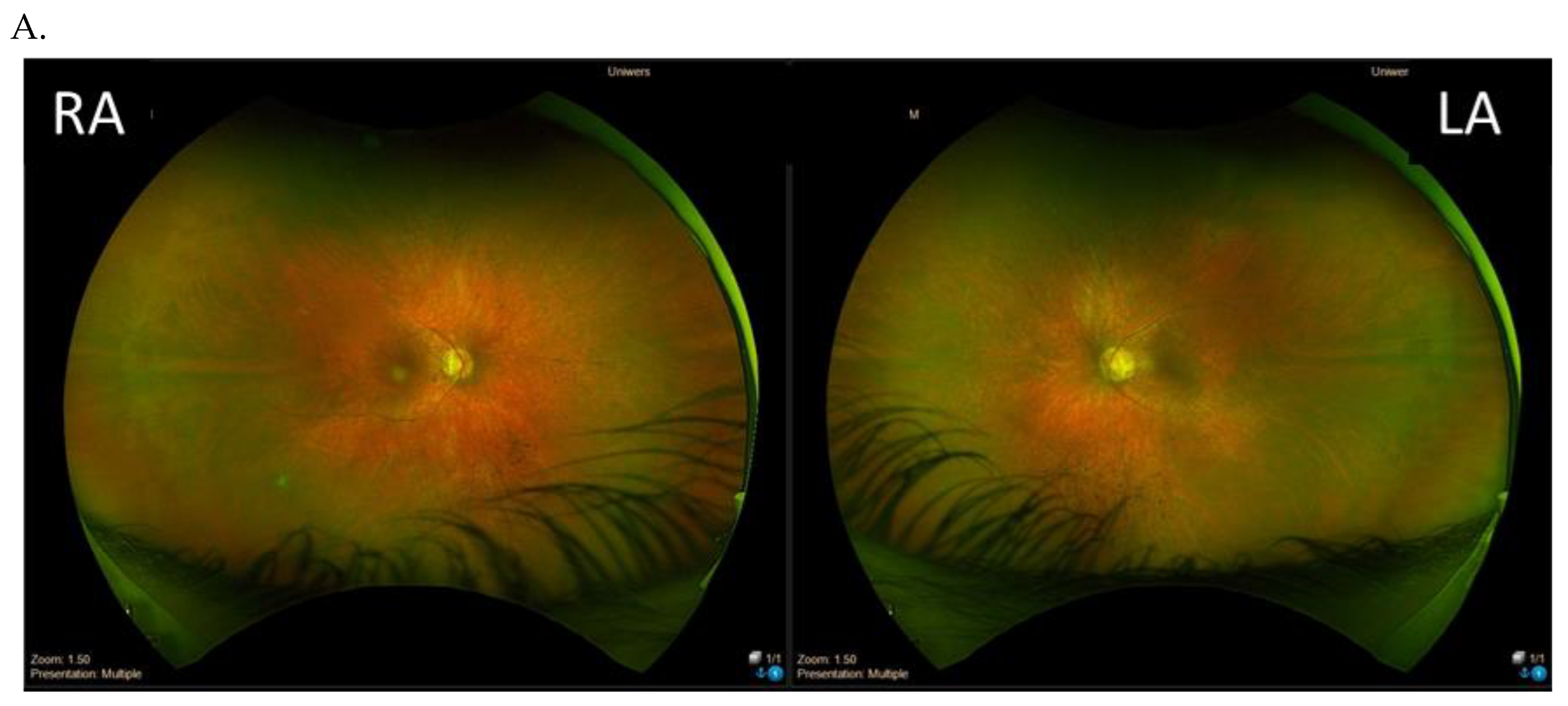{kind=link}
{kind=link}
| Patient ID | Family | Nucleotide Change | Exon | Classification | Class | Effect | Protein Change | Novelty | Reference |
|---|---|---|---|---|---|---|---|---|---|
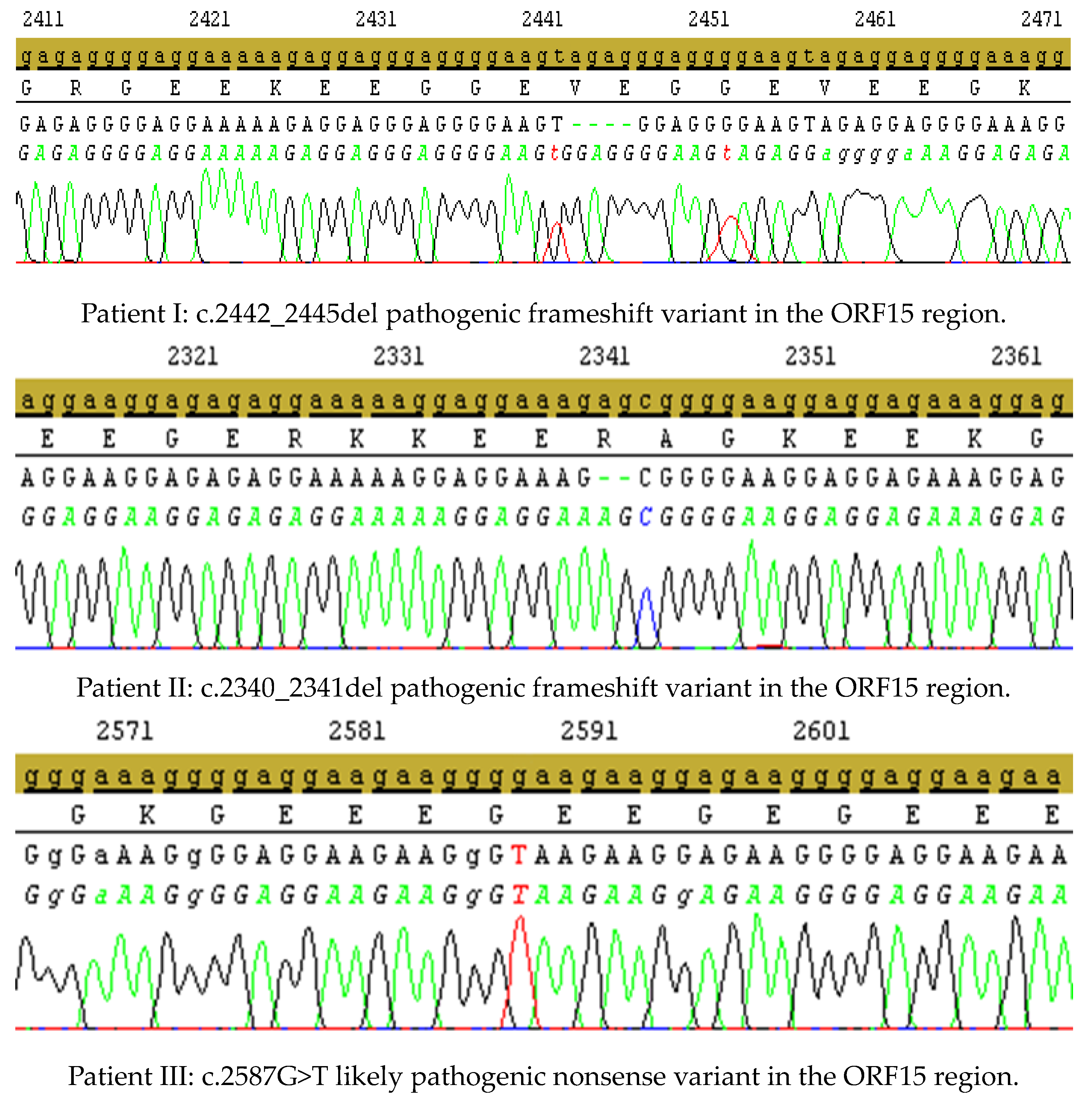
| I. | Family 1 | c.2442_2445del | ORF15 | deletion | pathogenic | frameshift | p.Gly817fs | no | Branham, 2012 [8] (PMID: 23150612), Vervoort, 2000 [5] (PMID: 10932196), Churchill, 2013 [19] (PMID: 23372056) |
| II. | Family 2 | c.2340_2341del | ORF15 | deletion | pathogenic | frameshift | p.Arg780fs | no | Bader, 2003 [7] (PMID: 12657579) Neidhardt, 2008 [11] (PMID: 18552978); |
| III. | Family 3 | c.2587G>T | ORF15 | SNV | likely pathogenic | nonsense | p.Glu863* | yes | this study |
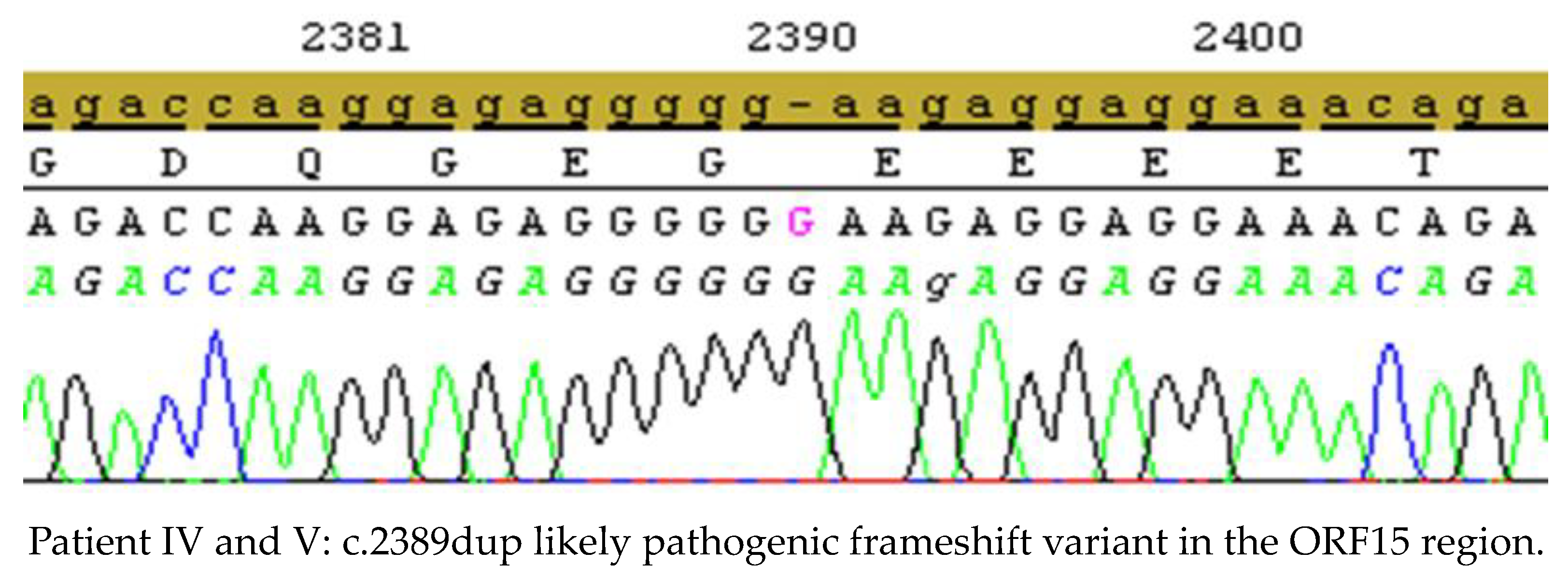
| IV. and V. | Family 4 | c.2389dup | ORF15 | duplication | likely pathogenic | frameshift | p.Glu797fs | yes | this study |
| VI. | Family 5 | c.2455dup | ORF15 | duplication | likely pathogenic | frameshift | p.Val819fs | yes | this study |
| VII. | Family 6 | c.593G>T | 6 | SNV | likely pathogenic | missense | p.Gly198Val- | yes | this study |
| VIII. | Family 7 | c.799G>C | 8 | SNV | likely pathogenic | missense | p.Gly267Arg- | yes | this study |
| Patient ID | Nucleotide Change | Age at Onset (Years) | Age at the Last Examination (Years) | Visual Acuity at the Examination (Snellen Charts) | Kinetic Visual Field | Optical Coherence Tomography (Central Foveal Thickness in Micrometers) |
|---|---|---|---|---|---|---|
| I (family 1) | c.2442_2445del | 10 | 31 | 0.7 0.7 | Advanced constriction, only V4e remarkable | 277 264 |
| II (family 2) | c.2340_2341del | 12 | 42 | 0.01 0.01 | Advanced constriction, only V4e remarkable | 208 205 |
| III (family 3) | c.2587G>T | 8 | 36 | 0.5 0.6 | Middle constriction, V4e, III4e and I4e remarkable | 143 147 |
| IV (family 4) | c.2389dup | 10 | 22 | 0.7 0.7 | Middle constriction, V4e, III4e remarkable | 223 225 |
| V (family 4) | c.2389dup | 10 | 22 | 0.4 0.2 | Middle constriction, V4e, III4e and I4e remarkable | 260 240 |
| VI (family 5) | c.2455dup | 14 | 25 | 0.6 0.3 | Middle constriction, V4e, remarkable | 205 186 |
| VII (family 6) | c.593G>T | 12 | 20 | 0.5 0.5 | Middle con-striction, V4e, III4e and I4e remarkable | 250 245 |
| VIII (family 7) | c.799G>C | 6 | 47 | 0.2 0.3 | Middle con-striction, V4e, III4e and I4e remarkable | 240 230 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nowomiejska, K.; Baltaziak, K.; Całka, P.; Ciesielka, M.; Teresiński, G.; Rejdak, R. Identification of the RPGR Gene Pathogenic Variants in a Cohort of Polish Male Patients with Retinitis Pigmentosa Phenotype. Genes 2023, 14, 1950. https://doi.org/10.3390/genes14101950
Nowomiejska K, Baltaziak K, Całka P, Ciesielka M, Teresiński G, Rejdak R. Identification of the RPGR Gene Pathogenic Variants in a Cohort of Polish Male Patients with Retinitis Pigmentosa Phenotype. Genes. 2023; 14(10):1950. https://doi.org/10.3390/genes14101950
Chicago/Turabian StyleNowomiejska, Katarzyna, Katarzyna Baltaziak, Paulina Całka, Marzanna Ciesielka, Grzegorz Teresiński, and Robert Rejdak. 2023. "Identification of the RPGR Gene Pathogenic Variants in a Cohort of Polish Male Patients with Retinitis Pigmentosa Phenotype" Genes 14, no. 10: 1950. https://doi.org/10.3390/genes14101950