
Role of RNA Alternative Splicing in T Cell Function and Disease

Abstract
:1. Introduction
2. Cell Surface Receptors
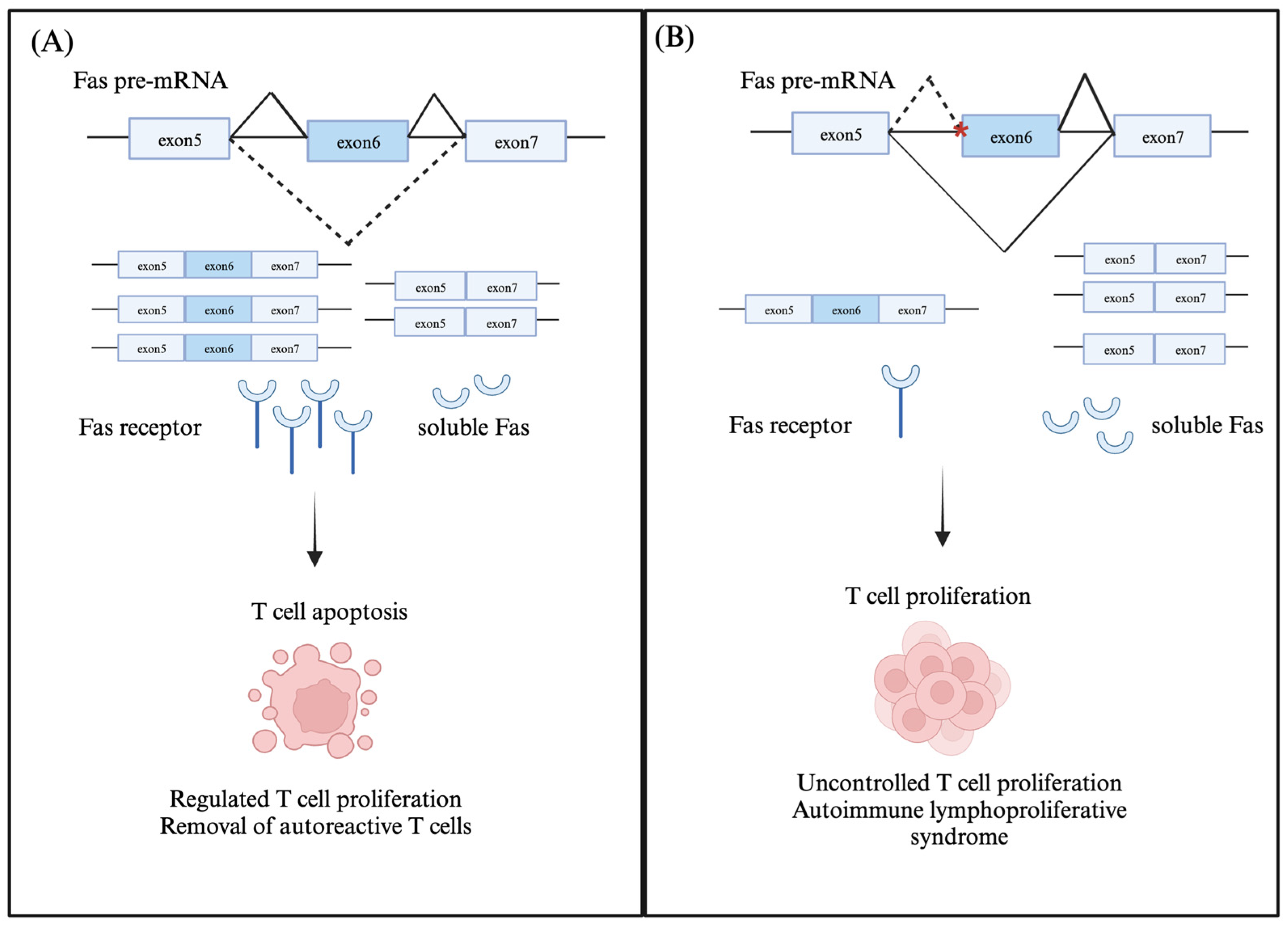
2.1. Fas (CD95/Apo-1)
2.2. Cytotoxic T lymphocyte Antigen 4 (CTLA-4)
3. Intracellular Signaling Factors
3.1. Lymphocyte-Specific Tyrosine Kinase (Lck)
3.2. CD3 Zeta Chain Associated 70kDa Protein (ZAP70)
4. Transcription Factors
Forkhead Transcription Factor 3 (FOXP3)
5. Factors Mediating Global Alternative Splicing Changes in T Cells
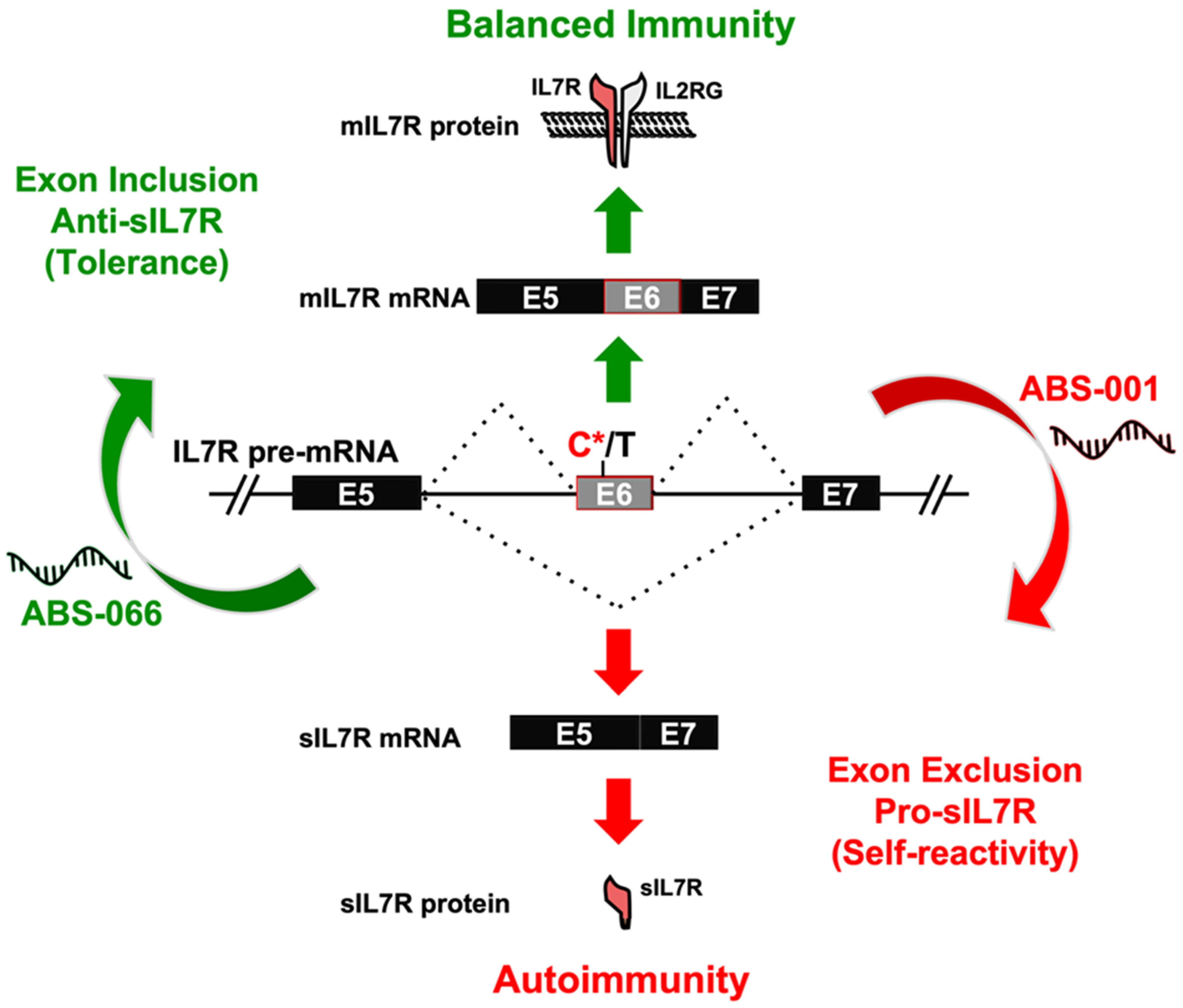
6. Therapeutic Modifications of Alternatively Spliced Variants in T Cells
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar, B.V.; Connors, T.J.; Farber, D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018, 48, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Wherry, E.J.; Ahmed, R. Effector and memory T-cell differentiation: Implications for vaccine development. Nat. Rev. Immunol. 2002, 2, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Josefowicz, S.Z.; Lu, L.-F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef] [PubMed]
- Martinez, N.M.; Lynch, K.W. Control of alternative splicing in immune responses: Many regulators, many predictions, much still to learn. Immunol. Rev. 2013, 253, 216–236. [Google Scholar] [CrossRef]
- Lynch, K.W. Consequences of regulated pre-mRNA splicing in the immune system. Nat. Rev. Immunol. 2004, 4, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.; Radens, C.M.; Ferretti, M.B.; Gazzara, M.R.; Lynch, K.W. Alternative splicing of apoptosis genes promotes human T cell survival. eLife 2022, 11, e80953. [Google Scholar] [CrossRef] [PubMed]
- Ip, J.Y.; Tong, A.; Pan, Q.; Topp, J.D.; Blencowe, B.J.; Lynch, K.W. Global analysis of alternative splicing during T-cell activation. RNA 2007, 13, 563–572. [Google Scholar] [CrossRef]
- Martinez, N.M.; Pan, Q.; Cole, B.S.; Yarosh, C.A.; Babcock, G.A.; Heyd, F.; Zhu, W.; Ajith, S.; Blencowe, B.J.; Lynch, K.W. Alternative splicing networks regulated by signaling in human T cells. RNA 2012, 18, 1029–1040. [Google Scholar] [CrossRef]
- Radens, C.M.; Blake, D.; Jewell, P.; Barash, Y.; Lynch, K.W. Meta-analysis of transcriptomic variation in T-cell populations reveals both variable and consistent signatures of gene expression and splicing. RNA 2020, 26, 1320–1333. [Google Scholar] [CrossRef]
- Blomhoff, A.; Kemp, E.H.; Gawkrodger, D.J.; Weetman, A.P.; Husebye, E.S.; Akselsen, H.E.; Lie, B.A.; Undlien, D.E. CTLA4 polymorphisms are associated with vitiligo, in patients with concomitant autoimmune diseases. Pigment Cell Res. 2005, 18, 55–58. [Google Scholar] [CrossRef]
- Evsyukova, I.; Somarelli, J.A.; Gregory, S.G.; Garcia-Blanco, M.A. Alternative splicing in multiple sclerosis and other autoimmune diseases. RNA Biol. 2010, 7, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Galarza-Muñoz, G.; Briggs, F.B.S.; Evsyukova, I.; Schott-Lerner, G.; Kennedy, E.M.; Nyanhete, T.; Wang, L.; Bergamaschi, L.; Widen, S.G.; Tomaras, G.D.; et al. Human Epistatic Interaction Controls IL7R Splicing and Increases Multiple Sclerosis Risk. Cell 2017, 169, 72–84.e13. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, E.; Kochanowska, I.E.; Frydecka, I.; Kiełbiński, M.; Potoczek, S.; Bilińska, M. The soluble CTLA-4 receptor: A new marker in autoimmune diseases. Arch. Immunol. Ther. Exp. 2005, 53, 336–341. [Google Scholar]
- Volpe, E.; Sambucci, M.; Battistini, L.; Borsellino, G. Fas-Fas Ligand: Checkpoint of T Cell Functions in Multiple Sclerosis. Front. Immunol. 2016, 7, 382. [Google Scholar] [CrossRef]
- Magerus, A.; Bercher-Brayer, C.; Rieux-Laucat, F. The genetic landscape of the FAS pathway deficiencies. Biomed. J. 2021, 44, 388–399. [Google Scholar] [CrossRef]
- van Doorn, R.; Dijkman, R.; Vermeer, M.H.; Starink, T.M.; Willemze, R.; Tensen, C.P. A Novel Splice Variant of the Fas Gene in Patients with Cutaneous T-Cell Lymphoma. Cancer Res. 2002, 62, 5389–5392. [Google Scholar]
- Tawara, M.; Maeda, T.; Yamada, Y.; Harasawa, H.; Tsuruda, K.; Sugahara, K.; Moriuchi, R.; Tomonaga, M.; Kamihira, S. Aberrant processing of Fas transcripts in adult T-cell leukemia: A possible role in tumor cell survival. Cancer Lett. 2003, 193, 235–242. [Google Scholar] [CrossRef]
- Choi, N.; Jang, H.N.; Oh, J.; Ha, J.; Park, H.; Zheng, X.; Lee, S.; Shen, H. SRSF6 Regulates the Alternative Splicing of the Apoptotic Fas Gene by Targeting a Novel RNA Sequence. Cancers 2022, 14, 1990. [Google Scholar] [CrossRef]
- Izquierdo, J.M. Hu antigen R (HuR) functions as an alternative pre-mRNA splicing regulator of Fas apoptosis-promoting receptor on exon definition. J. Biol. Chem. 2008, 283, 19077–19084. [Google Scholar] [CrossRef]
- Izquierdo, J.M. Cell-specific regulation of Fas exon 6 splicing mediated by Hu antigen R. Biochem. Biophys. Res. Commun. 2010, 402, 324–328. [Google Scholar] [CrossRef]
- Izquierdo, J.M. Heterogeneous ribonucleoprotein C displays a repressor activity mediated by T-cell intracellular antigen-1-related/like protein to modulate Fas exon 6 splicing through a mechanism involving Hu antigen R. Nucleic Acids Res. 2010, 38, 8001–8014. [Google Scholar] [CrossRef] [PubMed]
- Mickleburgh, I.; Kafasla, P.; Cherny, D.; Llorian, M.; Curry, S.; Jackson, R.J.; Smith, C.W.J. The organization of RNA contacts by PTB for regulation of FAS splicing. Nucleic Acids Res. 2014, 42, 8605–8620. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, L.; Mathur, R.; Braun, F.K.; Wise, J.F.; Berkova, Z.; Neelapu, S.; Kwak, L.W.; Samaniego, F. FAS-antisense 1 lncRNA and production of soluble versus membrane Fas in B-cell lymphoma. Leukemia 2014, 28, 2376–2387. [Google Scholar] [CrossRef] [PubMed]
- Agrebi, N.; Ben-Mustapha, I.; Matoussi, N.; Dhouib, N.; Ben-Ali, M.; Mekki, N.; Ben-Ahmed, M.; Larguèche, B.; Ben Becher, S.; Béjaoui, M.; et al. Rare Splicing Defects of FAS Underly Severe Recessive Autoimmune Lymphoproliferative Syndrome. Clin. Immunol. 2017, 183, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, H.; Posovszky, C.; Lahr, G.; van der Werff ten Bosch, J.; Boehler, T.; Debatin, K.-M. Residual CD95-pathway function in children with autoimmune lymphoproliferative syndrome is independent from clinical state and genotype of CD95 mutation. Pediatr. Res. 2009, 65, 163–168. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J. Exp. Med. 1996, 183, 2533–2540. [Google Scholar] [CrossRef]
- Gu, M.; Kakoulidou, M.; Giscombe, R.; Pirskanen, R.; Lefvert, A.K.; Klareskog, L.; Wang, X. Identification of CTLA-4 isoforms produced by alternative splicing and their association with myasthenia gravis. Clin. Immunol. 2008, 128, 374–381. [Google Scholar] [CrossRef]
- Kantarci, O.H.; Hebrink, D.D.; Achenbach, S.J.; Atkinson, E.J.; Waliszewska, A.; Buckle, G.; McMurray, C.T.; de Andrade, M.; Hafler, D.A.; Weinshenker, B.G. CTLA4 Is Associated with Susceptibility to Multiple Sclerosis. J. Neuroimmunol. 2003, 134, 133–141. [Google Scholar] [CrossRef]
- Dahal, L.N.; Basu, N.; Youssef, H.; Khanolkar, R.C.; Barker, R.N.; Erwig, L.P.; Ward, F.J. Immunoregulatory soluble CTLA-4 modifies effector T-cell responses in systemic lupus erythematosus. Arthritis Res. Ther. 2016, 18, 180. [Google Scholar] [CrossRef]
- Gerold, K.D.; Zheng, P.; Rainbow, D.B.; Zernecke, A.; Wicker, L.S.; Kissler, S. The soluble CTLA-4 splice variant protects from type 1 diabetes and potentiates regulatory T-cell function. Diabetes 2011, 60, 1955–1963. [Google Scholar] [CrossRef]
- Mahat, U.; Ambani, N.M.; Rotz, S.J.; Radhakrishnan, K. Heterozygous CTLA4 splice site mutation c.458-1G>C presenting with immunodeficiency and variable degree of immune dysregulation in three generation kindred of Caribbean descent. Pediatr. Hematol. Oncol. 2021, 38, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Rossy, J.; Williamson, D.; Gaus, K. How does the kinase Lck phosphorylate the T cell receptor? Spatial organization as a regulatory mechanism. Front. Immunol. 2012, 3, 167. [Google Scholar] [CrossRef] [PubMed]
- Dornan, S.; Sebestyen, Z.; Gamble, J.; Nagy, P.; Bodnar, A.; Alldridge, L.; Doe, S.; Holmes, N.; Goff, L.K.; Beverley, P.; et al. Differential association of CD45 isoforms with CD4 and CD8 regulates the actions of specific pools of p56lck tyrosine kinase in T cell antigen receptor signal transduction. J. Biol. Chem. 2002, 277, 1912–1918. [Google Scholar] [CrossRef] [PubMed]
- Horkova, V.; Drobek, A.; Paprckova, D.; Niederlova, V.; Prasai, A.; Uleri, V.; Glatzova, D.; Kraller, M.; Cesnekova, M.; Janusova, S.; et al. Unique roles of co-receptor-bound LCK in helper and cytotoxic T cells. Nat. Immunol. 2023, 24, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Palacios, E.H.; Weiss, A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 2004, 23, 7990–8000. [Google Scholar] [CrossRef]
- Kamens, J.S.; Ratnofsky, S.E.; Hirst, G.C. Lck Inhibitors as a Therapeutic Approach to Autoimmune Disease and Transplant Rejection. Curr. Opin. Investig. Drugs 2001, 2, 1213–1219. [Google Scholar]
- Kumar Singh, P.; Kashyap, A.; Silakari, O. Exploration of the Therapeutic Aspects of Lck: A Kinase Target in Inflammatory Mediated Pathological Conditions. Biomed. Pharmacother. 2018, 108, 1565–1571. [Google Scholar] [CrossRef]
- Germani, A.; Malherbe, S.; Rouer, E. The exon 7-spliced Lck isoform in T lymphocytes: A potential regulator of p56lck signaling pathways. Biochem. Biophys. Res. Commun. 2003, 301, 680–685. [Google Scholar] [CrossRef]
- Nervi, S.; Guinamard, R.; Delaval, B.; Lécine, P.; Vialettes, B.; Naquet, P.; Imbert, J. A rare mRNA variant of the human lymphocyte-specific protein tyrosine kinase LCK gene with intron B retention and exon 7 skipping encodes a putative protein with altered SH3-dependent molecular interactions. Gene 2005, 359, 18–25. [Google Scholar] [CrossRef]
- Rouer, E.; Benarous, R. Alternative splicing in the human lck gene leads to the deletion of exon 1’ and results in a new type II lck transcript. Oncogene 1992, 7, 2535–2538. [Google Scholar]
- Hulme, J.S.; Barratt, B.J.; Twells, R.C.J.; Cooper, J.D.; Lowe, C.E.; Howson, J.M.M.; Lam, A.C.; Smink, L.J.; Savage, D.A.; Undlien, D.E.; et al. Association analysis of the lymphocyte-specific protein tyrosine kinase (LCK) gene in type 1 diabetes. Diabetes 2004, 53, 2479–2482. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Wang, J.; Zhang, L.; Bian, W.; Lin, M.; Xu, X.; Zhou, X. LCK rs10914542-G allele associates with type 1 diabetes in children via T cell hyporesponsiveness. Pediatr. Res. 2019, 86, 311–315. [Google Scholar] [CrossRef]
- Wang, H.; Kadlecek, T.A.; Au-Yeung, B.B.; Goodfellow, H.E.S.; Hsu, L.Y.; Freedman, T.S.; Weiss, A. ZAP-70: An Essential Kinase in T-cell Signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002279. [Google Scholar] [CrossRef] [PubMed]
- Kuroyama, H.; Ikeda, T.; Kasai, M.; Yamasaki, S.; Tatsumi, M.; Utsuyama, M.; Saito, T.; Hirokawa, K. Identification of a novel isoform of ZAP-70, truncated ZAP kinase. Biochem. Biophys. Res. Commun. 2004, 315, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Picard, C.; Dogniaux, S.; Chemin, K.; Maciorowski, Z.; Lim, A.; Mazerolles, F.; Rieux-Laucat, F.; Stolzenberg, M.-C.; Debre, M.; Magny, J.-P.; et al. Hypomorphic Mutation of ZAP70 in Human Results in a Late Onset Immunodeficiency and No Autoimmunity. Eur. J. Immunol. 2009, 39, 1966–1976. [Google Scholar] [CrossRef]
- Hoshino, A.; Takashima, T.; Yoshida, K.; Morimoto, A.; Kawahara, Y.; Yeh, T.-W.; Okano, T.; Yamashita, M.; Mitsuiki, N.; Imai, K.; et al. Dysregulation of Epstein-Barr Virus Infection in Hypomorphic ZAP70 Mutation. J. Infect. Dis. 2018, 218, 825–834. [Google Scholar] [CrossRef]
- Josefowicz, S.Z.; Rudensky, A. Control of regulatory T cell lineage commitment and maintenance. Immunity 2009, 30, 616–625. [Google Scholar] [CrossRef]
- Konopacki, C.; Pritykin, Y.; Rubtsov, Y.; Leslie, C.S.; Rudensky, A.Y. Transcription factor Foxp1 regulates Foxp3 chromatin binding and coordinates regulatory T cell function. Nat. Immunol. 2019, 20, 232–242. [Google Scholar] [CrossRef]
- Plitas, G.; Rudensky, A.Y. Regulatory T Cells: Differentiation and Function. Cancer Immunol. Res. 2016, 4, 721–725. [Google Scholar] [CrossRef]
- Hirano, M.; Galarza-Muñoz, G.; Nagasawa, C.; Schott, G.; Wang, L.; Antonia, A.L.; Jain, V.; Yu, X.; Widen, S.G.; Briggs, F.B.S.; et al. The RNA helicase DDX39B activates FOXP3 RNA splicing to control T regulatory cell fate. eLife 2023, 12, e76927. [Google Scholar] [CrossRef]
- Mailer, R.K.W. Alternative Splicing of FOXP3—Virtue and Vice. Front. Immunol. 2018, 9, 530. [Google Scholar] [CrossRef] [PubMed]
- Mailer, R.K.W.; Joly, A.-L.; Liu, S.; Elias, S.; Tegner, J.; Andersson, J. IL-1β promotes Th17 differentiation by inducing alternative splicing of FOXP3. Sci. Rep. 2015, 5, 14674. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; West, K.O.; Sanghvi, S.; Laliotis, G.; Agosto, L.M.; Lynch, K.W.; Tsichlis, P.N.; Singh, H.; Patrick, K.L.; Guerau-de-Arellano, M. PRMT5 Promotes Symmetric Dimethylation of RNA Processing Proteins and Modulates Activated T Cell Alternative Splicing and Ca2+/NFAT Signaling. ImmunoHorizons 2021, 5, 884–897. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Nagai, Y.; Okumura, M.; Greene, M.I.; Kambayashi, T. PRMT5 Is Required for T Cell Survival and Proliferation by Maintaining Cytokine Signaling. Front. Immunol. 2020, 11, 621. [Google Scholar] [CrossRef]
- Corkery, D.P.; Holly, A.C.; Lahsaee, S.; Dellaire, G. Connecting the speckles: Splicing kinases and their role in tumorigenesis and treatment response. Nucleus 2015, 6, 279–288. [Google Scholar] [CrossRef]
- Gonçalves, V.; Henriques, A.F.A.; Pereira, J.F.S.; Neves Costa, A.; Moyer, M.P.; Moita, L.F.; Gama-Carvalho, M.; Matos, P.; Jordan, P. Phosphorylation of SRSF1 by SRPK1 regulates alternative splicing of tumor-related Rac1b in colorectal cells. RNA 2014, 20, 474–482. [Google Scholar] [CrossRef]
- Mathew, R.; Hartmuth, K.; Möhlmann, S.; Urlaub, H.; Ficner, R.; Lührmann, R. Phosphorylation of human PRP28 by SRPK2 is required for integration of the U4/U6-U5 tri-snRNP into the spliceosome. Nat. Struct. Mol. Biol. 2008, 15, 435–443. [Google Scholar] [CrossRef]
- Cassidy, M.F.; Herbert, Z.T.; Moulton, V.R. Splicing factor SRSF1 controls distinct molecular programs in regulatory and effector T cells implicated in systemic autoimmune disease. Mol. Immunol. 2022, 141, 94–103. [Google Scholar] [CrossRef]
- Katsuyama, T.; Moulton, V.R. Splicing factor SRSF1 is indispensable for regulatory T cell homeostasis and function. Cell Rep. 2021, 36, 109339. [Google Scholar] [CrossRef]
- Liu, Y.; Conaway, L.; Rutherford Bethard, J.; Al-Ayoubi, A.M.; Thompson Bradley, A.; Zheng, H.; Weed, S.A.; Eblen, S.T. Phosphorylation of the alternative mRNA splicing factor 45 (SPF45) by Clk1 regulates its splice site utilization, cell migration and invasion. Nucleic Acids Res. 2013, 41, 4949–4962. [Google Scholar] [CrossRef]
- McCuaig, R.D.; Dunn, J.; Li, J.; Masch, A.; Knaute, T.; Schutkowski, M.; Zerweck, J.; Rao, S. PKC-Theta is a Novel SC35 Splicing Factor Regulator in Response to T Cell Activation. Front. Immunol. 2015, 6, 562. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Y.; Xu, X.; Ding, J.-H.; Bermingham, J.R.; Fu, X.-D. SC35 Plays a Role in T Cell Development and Alternative Splicing of CD45. Mol. Cell 2001, 7, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Rogalska, M.E.; Vivori, C.; Valcárcel, J. Regulation of pre-mRNA splicing: Roles in physiology and disease, and therapeutic prospects. Nat. Rev. Genet. 2023, 24, 251–269. [Google Scholar] [CrossRef] [PubMed]
- Sako, Y.; Ninomiya, K.; Okuno, Y.; Toyomoto, M.; Nishida, A.; Koike, Y.; Ohe, K.; Kii, I.; Yoshida, S.; Hashimoto, N.; et al. Development of an orally available inhibitor of CLK1 for skipping a mutated dystrophin exon in Duchenne muscular dystrophy. Sci. Rep. 2017, 7, 46126. [Google Scholar] [CrossRef]
- Shibata, S.; Ajiro, M.; Hagiwara, M. Mechanism-Based Personalized Medicine for Cystic Fibrosis by Suppressing Pseudo Exon Inclusion. Cell Chem Biol. 2020, 27, 1472–1482.e6. [Google Scholar] [CrossRef]
- Webb, L.M.; Amici, S.A.; Jablonski, K.A.; Savardekar, H.; Panfil, A.R.; Li, L.; Zhou, W.; Peine, K.; Karkhanis, V.; Bachelder, E.M.; et al. PRMT5-Selective Inhibitors Suppress Inflammatory T Cell Responses and Experimental Autoimmune Encephalomyelitis. J. Immunol. Baltim. Md 1950 2017, 198, 1439–1451. [Google Scholar] [CrossRef]
- Xu, C.; Chen, X.; Zhang, X.; Zhao, D.; Dou, Z.; Xie, X.; Li, H.; Yang, H.; Li, Q.; Zhang, H.; et al. RNA-binding protein 39: A promising therapeutic target for cancer. Cell Death Discov. 2021, 7, 214. [Google Scholar] [CrossRef]
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef]
- Charleston, J.S.; Schnell, F.J.; Dworzak, J.; Donoghue, C.; Lewis, S.; Chen, L.; Young, G.D.; Milici, A.J.; Voss, J.; DeAlwis, U.; et al. Eteplirsen treatment for Duchenne muscular dystrophy: Exon skipping and dystrophin production. Neurology 2018, 90, e2146–e2154. [Google Scholar] [CrossRef]
- Cirak, S.; Arechavala-Gomeza, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; Wells, D.J.; et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet 2011, 378, 595–605. [Google Scholar] [CrossRef]
- Frank, D.E.; Schnell, F.J.; Akana, C.; El-Husayni, S.H.; Desjardins, C.A.; Morgan, J.; Charleston, J.S.; Sardone, V.; Domingos, J.; Dickson, G.; et al. Increased Dystrophin Production With Golodirsen in Patients With Duchenne Muscular Dystrophy. Neurology. 2023, 100, 936. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Y.; Chang, G.; Wang, F.; Wang, F.; Geng, X. Alternative Splicing of hTERT Pre-mRNA: A Potential Strategy for the Regulation of Telomerase Activity. Int. J. Mol. Sci. 2017, 18, 567. [Google Scholar] [CrossRef] [PubMed]
- Galarza-Muñoz, G.; Kennedy-Boone, D.; Schott, G.; Bradrick, S.S.; Garcia-Blanco, M.A. Antisense modulation of IL7R splicing to control sIL7R expression in human CD4+ T cells. RNA 2022, 28, 1058–1073. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | Gene Description | Splice Site Mutation | Genomic Region | Condition(s) |
|---|---|---|---|---|
| CTLA4 | cytotoxic T-lymphocyte-associated protein 4 | NM_005214.5(CTLA4):c.458-1G>T | Intron 2 | Autoimmune lymphoproliferative syndrome due to CTLA4 haploinsuffiency |
| NM_005214.5(CTLA4):c.458-1G>C | Intron 2 | |||
| FAS | Fas cell surface death receptor | NM_000043.6(FAS):c.506-1G>C | Intron 5 | Autoimmune lymphoproliferative syndrome type 1 |
| NM_000043.6(FAS):c.334+2T>C | Intron 3 | |||
| NM_000043.6(FAS):c.334+2dup | Intron 3 | |||
| NM_000043.6(FAS):c.335-2A>G | Intron 3 | |||
| NM_000043.6(FAS):c.569-2A>C | Intron 6 | |||
| NM_000043.6(FAS):c.651+1G>A | Intron 7 | |||
| NM_000043.6(FAS):c.651+1G>T | Intron 7 | |||
| NM_000043.6(FAS):c.651+2T>A | Intron 7 | |||
| NM_000043.6(FAS):c.651+2T>C | Intron 7 | |||
| NM_000043.6(FAS):c.651+2_651+3insTGAAAT | Intron 7 | |||
| NM_000043.6(FAS):c.652-1G>A | Intron 7 | |||
| NM_000043.6(FAS):c.676+1G>A | Intron 8 | |||
| NM_000043.6(FAS):c.676+1G>C | Intron 8 | |||
| NM_000043.6(FAS):c.676+1G>T | Intron 8 | |||
| FOXP3 | forkhead box P3 | NM_014009.4(FOXP3):c.736-2A>T | Intron 7 | Insulin-dependent diabetes mellitus, Secretory diarrhea syndrome |
| NM_014009.4(FOXP3):c.-23+1G>T | Exon1-intron1 | |||
| NM_014009.4(FOXP3):c.210+1G>A | Intron 2 | |||
| NM_014009.4(FOXP3):c.210+1G>T | Intron 2 | |||
| IL7R | interleukin 7 receptor | NM_002185.5(IL7R):c.221+2T>G | Intron 2 | Immunodeficiency 104, Severe combined immunodeficiency syndrome |
| NM_002185.5(IL7R):c.221+1G>A | Intron 2 | |||
| NM_002185.5(IL7R):c.537+1G>A | Intron 4 | |||
| NM_002185.5(IL7R):c.[134A>C;537+1G>A] | Intron 4 | |||
| NM_002185.5(IL7R):c.707-2A>G | Intron 5 | |||
| NM_002185.5(IL7R):c.83-1G>A | Intron 1 | |||
| NM_002185.5(IL7R):c.221+2T>G | Intron 2 | |||
| NM_002185.5(IL7R):c.538-1G>A | Intron 4 | |||
| LCK | LCK proto-oncogene, Src family tyrosine kinase | NM_005356.5(LCK):c.481+2T>G | Intron 6 | Severe combined immunodeficiency due to LCK deficiency |
| ZAP70 | zeta chain of T-cell-receptor-associated protein kinase 70 | NM_001079.4(ZAP70):c.703-1G>A | Intron 5 | ZAP70-related severe combined immunodeficiency |
| NM_001079.4(ZAP70):c.791-1G>A | Intron 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banerjee, S.; Galarza-Muñoz, G.; Garcia-Blanco, M.A. Role of RNA Alternative Splicing in T Cell Function and Disease. Genes 2023, 14, 1896. https://doi.org/10.3390/genes14101896
Banerjee S, Galarza-Muñoz G, Garcia-Blanco MA. Role of RNA Alternative Splicing in T Cell Function and Disease. Genes. 2023; 14(10):1896. https://doi.org/10.3390/genes14101896
Chicago/Turabian StyleBanerjee, Shefali, Gaddiel Galarza-Muñoz, and Mariano A. Garcia-Blanco. 2023. "Role of RNA Alternative Splicing in T Cell Function and Disease" Genes 14, no. 10: 1896. https://doi.org/10.3390/genes14101896