
Occult Macular Dysfunction Syndrome: Identification of Multiple Pathologies in a Clinical Spectrum of Macular Dysfunction with Normal Fundus in East Asian Patients: EAOMD Report No. 5

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Clinical Investigation
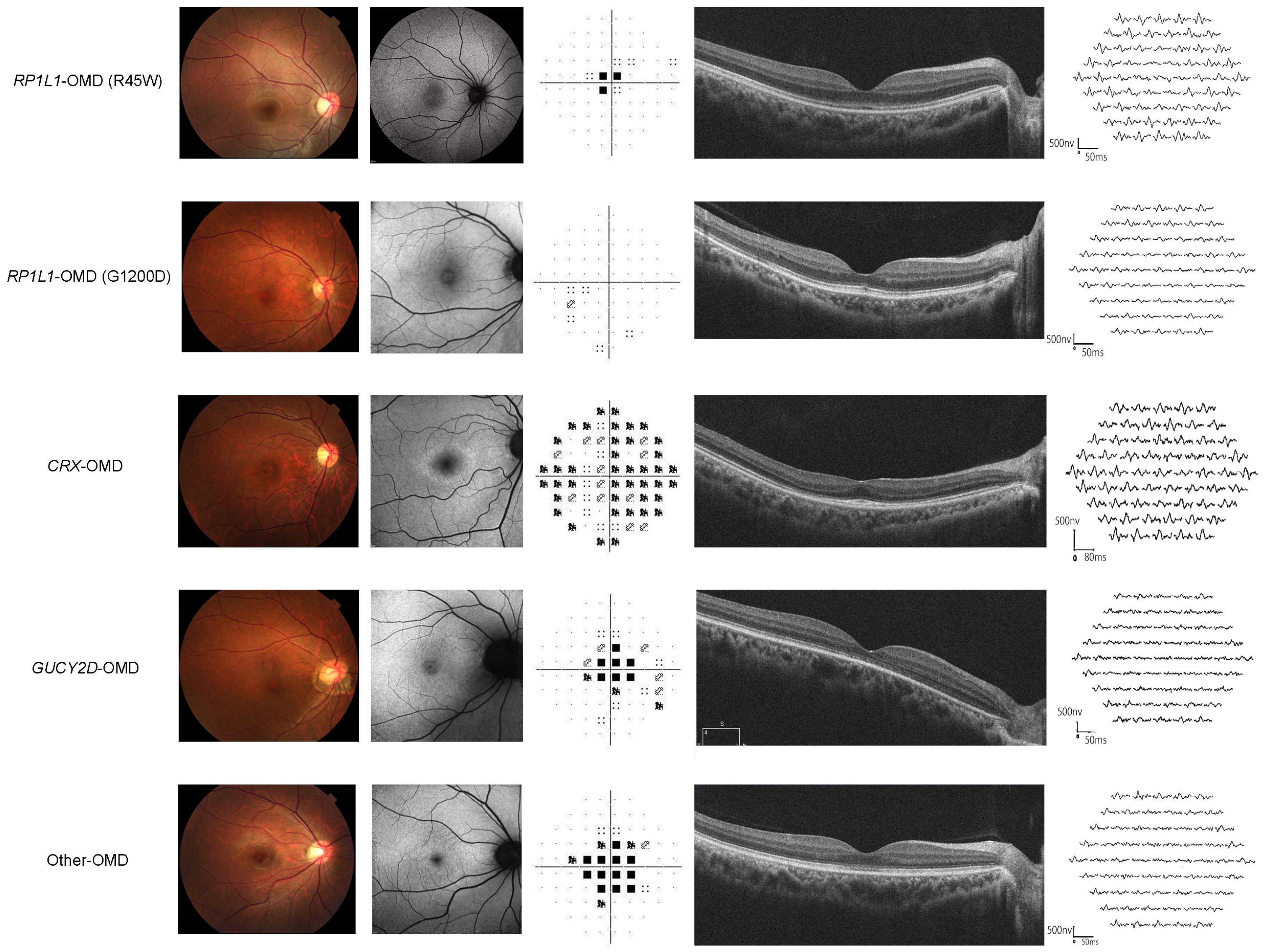
2.3. Classification of Clinical Parameters (VF, mfERG, SD-OCT)
2.4. Molecular Genetic Analysis
2.5. Genotype Subgroup Classification
2.6. Comparison of Clinical Parameters and Clinical Classifications (VF/mfERG/SD-OCT)
2.7. Statistical Analysis
3. Results
3.1. Patients
3.2. Demographics and Clinical Findings
3.3. Classification of Clinical Parameters (VF, mfERG, SD-OCT)
3.4. Molecular Genetics
3.5. Demographics for Each Genotype Group
3.6. Clinical Parameters and Classifications for Each Genotype Group
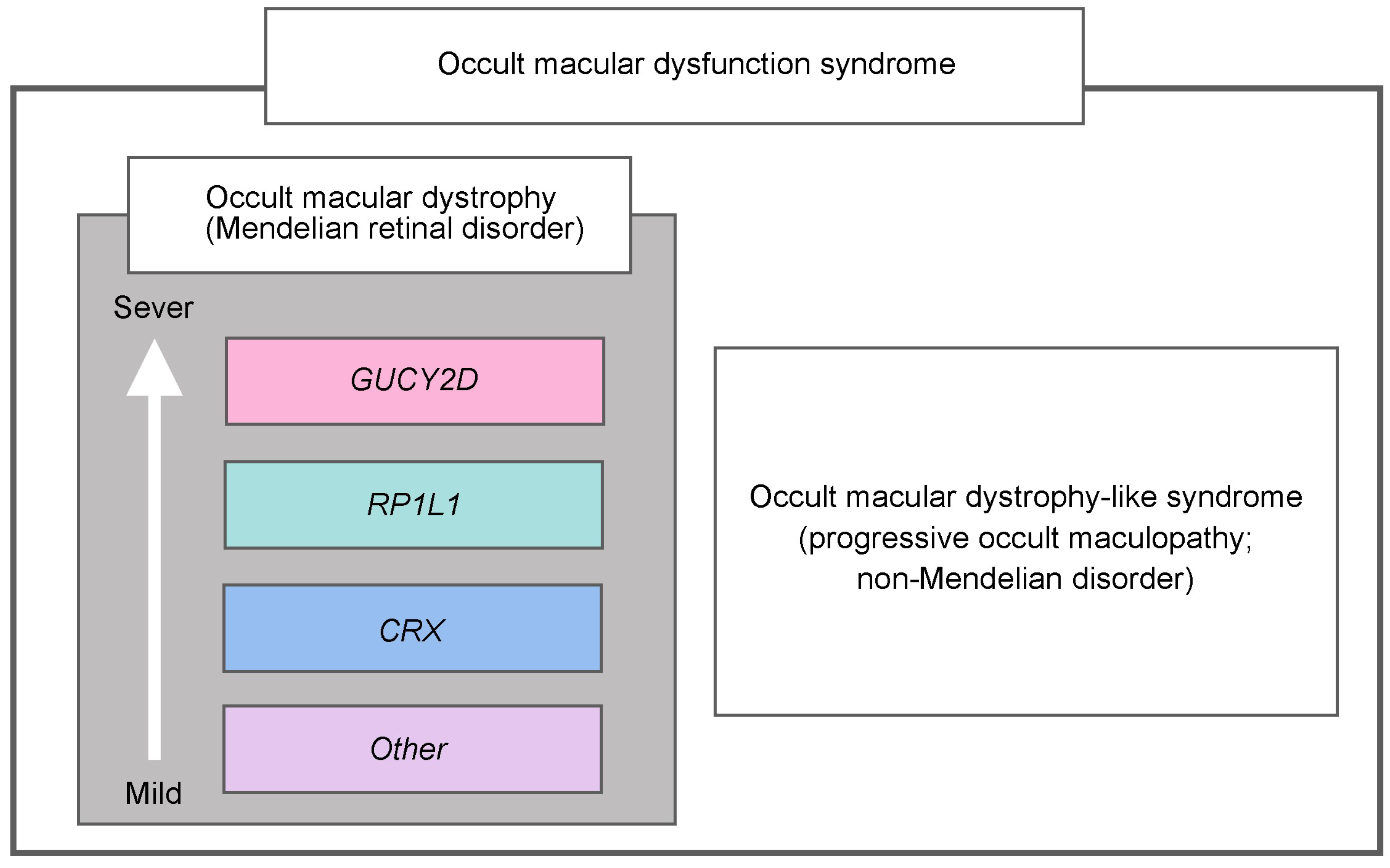
3.7. Comparison Analyses among Genotype Groups
3.8. Genotype R+ Data Set
3.9. Kaplan-Meier Survival Analyses for BCVA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miyake, Y.; Ichikawa, K.; Shiose, Y.; Kawase, Y. Hereditary macular dystrophy without visible fundus abnormality. Am. J. Ophthalmol. 1989, 108, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Horiguchi, M.; Tomita, N.; Kondo, M.; Tanikawa, A.; Takahashi, H.; Suzuki, S.; Terasaki, H. Occult macular dystrophy. Am. J. Ophthalmol. 1996, 122, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Tsunoda, K. Occult macular dystrophy. Jpn. J. Ophthalmol. 2015, 59, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Fujinami-Yokokawa Y, R.A.; Sergouniotis, P.I.; Fujinaim, K. Occult macular dystrophy. Clin. Ophthalmic Genet. Genom. 2022, 1, 241–245. [Google Scholar]
- Noel, N.C.L.; MacDonald, I.M. RP1L1 and inherited photoreceptor disease: A review. Surv. Ophthalmol. 2020, 65, 725–739. [Google Scholar] [CrossRef]
- Fujinami, K.; Yang, L.; Joo, K.; Tsunoda, K.; Kameya, S.; Hanazono, G.; Fujinami-Yokokawa, Y.; Arno, G.; Kondo, M.; Nakamura, N.; et al. Clinical and Genetic Characteristics of East Asian Patients with Occult Macular Dystrophy (Miyake Disease): East Asia Occult Macular Dystrophy Studies Report Number 1. Ophthalmology 2019, 126, 1432–1444. [Google Scholar] [CrossRef]
- Yang, L.; Joo, K.; Tsunoda, K.; Kondo, M.; Fujinami-Yokokawa, Y.; Arno, G.; Pontikos, N.; Liu, X.; Nakamura, N.; Kurihara, T.; et al. Spatial Functional Characteristics of East Asian Patients with Occult Macular Dystrophy (Miyake Disease); EAOMD Report No. 2. Am. J. Ophthalmol. 2021, 221, 169–180. [Google Scholar] [CrossRef]
- Ahn, S.J.; Yang, L.; Tsunoda, K.; Kondo, M.; Fujinami-Yokokawa, Y.; Nakamura, N.; Iwata, T.; Kim, M.S.; Mun, Y.; Park, J.Y.; et al. Visual Field Characteristics in East Asian Patients with Occult Macular Dystrophy (Miyake Disease): EAOMD Report No. 3. Invest. Ophthalmol. Vis. Sci. 2022, 63, 12. [Google Scholar] [CrossRef]
- Fujinami, K.; Tsunoda, K.; Hanazono, G.; Shinoda, K.; Ohde, H.; Miyake, Y. Fundus autofluorescence in autosomal dominant occult macular dystrophy. Arch. Ophthalmol. 2011, 129, 597–602. [Google Scholar] [CrossRef]
- Ahn, S.J.; Cho, S.I.; Ahn, J.; Park, S.S.; Park, K.H.; Woo, S.J. Clinical and genetic characteristics of Korean occult macular dystrophy patients. Investig. Ophthalmol. Vis. Sci. 2013, 54, 4856–4863. [Google Scholar] [CrossRef]
- Fujinami, K.; Kameya, S.; Kikuchi, S.; Ueno, S.; Kondo, M.; Hayashi, T.; Shinoda, K.; Machida, S.; Kuniyoshi, K.; Kawamura, Y.; et al. Novel RP1L1 Variants and Genotype-Photoreceptor Microstructural Phenotype Associations in Cohort of Japanese Patients with Occult Macular Dystrophy. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4837–4846. [Google Scholar] [CrossRef] [PubMed]
- Suga, A.; Yoshitake, K.; Minematsu, N.; Tsunoda, K.; Fujinami, K.; Miyake, Y.; Kuniyoshi, K.; Hayashi, T.; Mizobuchi, K.; Ueno, S.; et al. Genetic characterization of 1210 Japanese pedigrees with inherited retinal diseases by whole-exome sequencing. Hum. Mutat. 2022, 43, 2251–2264. [Google Scholar] [CrossRef] [PubMed]
- Luoma-Overstreet, G.; Jewell, A.; Brar, V.; Couser, N. Occult Macular Dystrophy: A case report and major review. Ophthalmic Genet. 2022, 43, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chen, K.J.; Lai, C.C.; Wu, W.C.; Wang, N.K. Clinical Features in a Case of Occult Macular Dystrophy with Rp1l1 Mutation. Retin. Cases Brief. Rep. 2019, 13, 158–161. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Gao, F.J.; Li, J.K.; Chen, F.; Hu, F.Y.; Xu, G.Z.; Zhang, J.G.; Sun, H.X.; Zhang, S.H.; Xu, P.; et al. Clinical and Genetic Characteristics of Chinese Patients with Occult Macular Dystrophy. Investig. Ophthalmol. Vis. Sci. 2020, 61, 10. [Google Scholar] [CrossRef]
- Kim, M.S.; Joo, K.; Seong, M.W.; Kim, M.J.; Park, K.H.; Park, S.S.; Woo, S.J. Genetic Mutation Profiles in Korean Patients with Inherited Retinal Diseases. J. Korean Med. Sci. 2019, 34, e161. [Google Scholar] [CrossRef]
- Fujii, S.; Escano, M.F.; Ishibashi, K.; Matsuo, H.; Yamamoto, M. Multifocal electroretinography in patients with occult macular dystrophy. Br. J. Ophthalmol. 1999, 83, 879–880. [Google Scholar] [CrossRef]
- Piao, C.H.; Kondo, M.; Tanikawa, A.; Terasaki, H.; Miyake, Y. Multifocal electroretinogram in occult macular dystrophy. Investig. Ophthalmol. Vis. Sci. 2000, 41, 513–517. [Google Scholar]
- Davidson, A.E.; Sergouniotis, P.I.; Mackay, D.S.; Wright, G.A.; Waseem, N.H.; Michaelides, M.; Holder, G.E.; Robson, A.G.; Moore, A.T.; Plagnol, V.; et al. RP1L1 variants are associated with a spectrum of inherited retinal diseases including retinitis pigmentosa and occult macular dystrophy. Hum. Mutat. 2013, 34, 506–514. [Google Scholar] [CrossRef]
- Wildberger, H.; Niemeyer, G.; Junghardt, A. Multifocal electroretinogram (mfERG) in a family with occult macular dystrophy (OMD). Klin. Monbl Augenheilkd. 2003, 220, 111–115. [Google Scholar] [CrossRef]
- Kondo, M.; Ueno, S.; Piao, C.H.; Ito, Y.; Terasaki, H.; Miyake, Y. Occult macular dystrophy in an 11 year old boy. Br. J. Ophthalmol. 2004, 88, 1602–1603. [Google Scholar] [CrossRef] [PubMed]
- Okuno, T.; Oku, H.; Kondo, M.; Miyake, Y.; Sugasawa, J.; Utsumi, T.; Ikeda, T. Abnormalities of visual-evoked potentials and pupillary light reflexes in a family with autosomal dominant occult macular dystrophy. Clin. Exp. Ophthalmol. 2007, 35, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Hanazono, G.; Ohde, H.; Shinoda, K.; Tsunoda, K.; Tsubota, K.; Miyake, Y. Pattern-reversal visual-evoked potential in patients with occult macular dystrophy. Clin. Ophthalmol. 2010, 4, 1515–1520. [Google Scholar] [CrossRef]
- Park, S.J.; Woo, S.J.; Park, K.H.; Hwang, J.M.; Chung, H. Morphologic photoreceptor abnormality in occult macular dystrophy on spectral-domain optical coherence tomography. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3673–3679. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, K.; Usui, T.; Hatase, T.; Yamai, S.; Fujinami, K.; Hanazono, G.; Shinoda, K.; Ohde, H.; Akahori, M.; Iwata, T.; et al. Clinical characteristics of occult macular dystrophy in family with mutation of RP1l1 gene. Retina 2012, 32, 1135–1147. [Google Scholar] [CrossRef]
- Kato, Y.; Hanazono, G.; Fujinami, K.; Hatase, T.; Kawamura, Y.; Iwata, T.; Miyake, Y.; Tsunoda, K. Parafoveal Photoreceptor Abnormalities in Asymptomatic Patients with RP1L1 Mutations in Families with Occult Macular Dystrophy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 6020–6029. [Google Scholar] [CrossRef]
- Fujinami-Yokokawa, Y.; Pontikos, N.; Yang, L.; Tsunoda, K.; Yoshitake, K.; Iwata, T.; Miyata, H.; Fujinami, K.; Japan Eye Genetics Consortium, O.B.O. Prediction of Causative Genes in Inherited Retinal Disorders from Spectral-Domain Optical Coherence Tomography Utilizing Deep Learning Techniques. J. Ophthalmol. 2019, 2019, 1691064. [Google Scholar] [CrossRef]
- Nakamura, N.; Tsunoda, K.; Mizuno, Y.; Usui, T.; Hatase, T.; Ueno, S.; Kuniyoshi, K.; Hayashi, T.; Katagiri, S.; Kondo, M.; et al. Clinical Stages of Occult Macular Dystrophy Based on Optical Coherence Tomographic Findings. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4691–4700. [Google Scholar] [CrossRef]
- Tsunoda, K.; Hanazono, G. Detailed analyses of microstructure of photoreceptor layer at different severities of occult macular dystrophy by ultrahigh-resolution SD-OCT. Am. J. Ophthalmol. Case Rep. 2022, 26, 101490. [Google Scholar] [CrossRef]
- Zobor, D.; Zobor, G.; Hipp, S.; Baumann, B.; Weisschuh, N.; Biskup, S.; Sliesoraityte, I.; Zrenner, E.; Kohl, S. Phenotype Variations Caused by Mutations in the RP1L1 Gene in a Large Mainly German Cohort. Investig. Ophthalmol. Vis. Sci. 2018, 59, 3041–3052. [Google Scholar] [CrossRef]
- Takahashi, H.; Hayashi, T.; Tsuneoka, H.; Nakano, T.; Yamada, H.; Katagiri, S.; Fujino, Y.; Noda, Y.; Yoshimoto, M.; Kawashima, H. Occult macular dystrophy with bilateral chronic subfoveal serous retinal detachment associated with a novel RP1L1 mutation (p.S1199P). Doc. Ophthalmol. 2014, 129, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Kabuto, T.; Takahashi, H.; Goto-Fukuura, Y.; Igarashi, T.; Akahori, M.; Kameya, S.; Iwata, T.; Mizota, A.; Yamaki, K.; Miyake, Y.; et al. A new mutation in the RP1L1 gene in a patient with occult macular dystrophy associated with a depolarizing pattern of focal macular electroretinograms. Mol. Vis. 2012, 18, 1031–1039. [Google Scholar] [PubMed]
- Akahori, M.; Tsunoda, K.; Miyake, Y.; Fukuda, Y.; Ishiura, H.; Tsuji, S.; Usui, T.; Hatase, T.; Nakamura, M.; Ohde, H.; et al. Dominant mutations in RP1L1 are responsible for occult macular dystrophy. Am. J. Hum. Genet. 2010, 87, 424–429. [Google Scholar] [CrossRef]
- Fujinami-Yokokawa, Y.; Ninomiya, H.; Liu, X.; Yang, L.; Pontikos, N.; Yoshitake, K.; Iwata, T.; Sato, Y.; Hashimoto, T.; Tsunoda, K.; et al. Prediction of causative genes in inherited retinal disorder from fundus photography and autofluorescence imaging using deep learning techniques. Br. J. Ophthalmol. 2021, 105, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.J.; Ahn, J.; Park, K.H.; Woo, S.J. Multimodal imaging of occult macular dystrophy. JAMA Ophthalmol. 2013, 131, 880–890. [Google Scholar] [CrossRef]
- Koyanagi, Y.; Akiyama, M.; Nishiguchi, K.M.; Momozawa, Y.; Kamatani, Y.; Takata, S.; Inai, C.; Iwasaki, Y.; Kumano, M.; Murakami, Y.; et al. Genetic characteristics of retinitis pigmentosa in 1204 Japanese patients. J. Med. Genet. 2019, 56, 662–670. [Google Scholar] [CrossRef]
- Oishi, M.; Oishi, A.; Gotoh, N.; Ogino, K.; Higasa, K.; Iida, K.; Makiyama, Y.; Morooka, S.; Matsuda, F.; Yoshimura, N. Comprehensive molecular diagnosis of a large cohort of Japanese retinitis pigmentosa and Usher syndrome patients by next-generation sequencing. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7369–7375. [Google Scholar] [CrossRef]
- Hu, Y.S.; Song, H.; Li, Y.; Xiao, Z.Y.; Li, T. Whole-exome sequencing identifies novel mutations in genes responsible for retinitis pigmentosa in 2 nonconsanguineous Chinese families. Int. J. Ophthalmol. 2019, 12, 915–923. [Google Scholar] [CrossRef]
- Fujinami-Yokokawa, Y.; Fujinami, K.; Kuniyoshi, K.; Hayashi, T.; Ueno, S.; Mizota, A.; Shinoda, K.; Arno, G.; Pontikos, N.; Yang, L.; et al. Clinical and Genetic Characteristics of 18 Patients from 13 Japanese Families with CRX-associated retinal disorder: Identification of Genotype-phenotype Association. Sci. Rep. 2020, 10, 9531. [Google Scholar] [CrossRef]
- Liu, X.; Fujinami, K.; Kuniyoshi, K.; Kondo, M.; Ueno, S.; Hayashi, T.; Mochizuki, K.; Kameya, S.; Yang, L.; Fujinami-Yokokawa, Y.; et al. Clinical and Genetic Characteristics of 15 Affected Patients From 12 Japanese Families with GUCY2D-Associated Retinal Disorder. Transl. Vis. Sci. Technol. 2020, 9, 2. [Google Scholar] [CrossRef]
- Hood, D.C.; Bach, M.; Brigell, M.; Keating, D.; Kondo, M.; Lyons, J.S.; Marmor, M.F.; McCulloch, D.L.; Palmowski-Wolfe, A.M.; International Society For Clinical Electrophysiology of Vision. ISCEV standard for clinical multifocal electroretinography (mfERG) (2011 edition). Doc. Ophthalmol. 2012, 124, 1–13. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.B.; Bach, M.; Kondo, M.; Li, S.; Walker, S.; Holopigian, K.; Viswanathan, S.; Robson, A.G. ISCEV standard for clinical multifocal electroretinography (mfERG) (2021 update). Doc. Ophthalmol. 2021, 142, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Robson, A.G.; Frishman, L.J.; Grigg, J.; Hamilton, R.; Jeffrey, B.G.; Kondo, M.; Li, S.; McCulloch, D.L. ISCEV Standard for full-field clinical electroretinography (2022 update). Doc. Ophthalmol. 2022, 144, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, K.; Oishi, A.; Yang, L.; Arno, G.; Pontikos, N.; Yoshitake, K.; Fujinami-Yokokawa, Y.; Liu, X.; Hayashi, T.; Katagiri, S.; et al. Clinical and genetic characteristics of 10 Japanese patients with PROM1-associated retinal disorder: A report of the phenotype spectrum and a literature review in the Japanese population. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 656–674. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M.; ClinGen Sequence Variant Interpretation Working, G. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef]
- Payne, A.M.; Morris, A.G.; Downes, S.M.; Johnson, S.; Bird, A.C.; Moore, A.T.; Bhattacharya, S.S.; Hunt, D.M. Clustering and frequency of mutations in the retinal guanylate cyclase (GUCY2D) gene in patients with dominant cone-rod dystrophies. J. Med. Genet. 2001, 38, 611–614. [Google Scholar] [CrossRef]
- de Castro-Miro, M.; Pomares, E.; Lores-Motta, L.; Tonda, R.; Dopazo, J.; Marfany, G.; Gonzalez-Duarte, R. Combined genetic and high-throughput strategies for molecular diagnosis of inherited retinal dystrophies. PLoS ONE 2014, 9, e88410. [Google Scholar] [CrossRef]
- Ito, S.; Nakamura, M.; Ohnishi, Y.; Miyake, Y. Autosomal dominant cone-rod dystrophy with R838H and R838C mutations in the GUCY2D gene in Japanese patients. Jpn. J. Ophthalmol. 2004, 48, 228–235. [Google Scholar] [CrossRef]
- Ito, S.; Nakamura, M.; Nuno, Y.; Ohnishi, Y.; Nishida, T.; Miyake, Y. Novel complex GUCY2D mutation in Japanese family with cone-rod dystrophy. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Weigell-Weber, M.; Fokstuen, S.; Torok, B.; Niemeyer, G.; Schinzel, A.; Hergersberg, M. Codons 837 and 838 in the retinal guanylate cyclase gene on chromosome 17p: Hot spots for mutations in autosomal dominant cone-rod dystrophy? Arch Ophthalmol 2000, 118, 300. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Xu, K.; Zhang, X.; Xie, Y.; Bai, F.; Li, Y. GUCY2D mutations in a Chinese cohort with autosomal dominant cone or cone-rod dystrophies. Doc. Ophthalmol. 2015, 131, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Kitiratschky, V.B.; Wilke, R.; Renner, A.B.; Kellner, U.; Vadala, M.; Birch, D.G.; Wissinger, B.; Zrenner, E.; Kohl, S. Mutation analysis identifies GUCY2D as the major gene responsible for autosomal dominant progressive cone degeneration. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5015–5023. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.; Robson, A.G.; Holder, G.E.; Stockman, A.; Egan, C.A.; Moore, A.T.; Webster, A.R. A detailed phenotypic description of autosomal dominant cone dystrophy due to a de novo mutation in the GUCY2D gene. Eye 2014, 28, 481–487. [Google Scholar] [CrossRef]
- Rodilla, C.; Martin-Merida, I.; Blanco-Kelly, F.; Trujillo-Tiebas, M.J.; Avila-Fernandez, A.; Riveiro-Alvarez, R.; Del Pozo-Valero, M.; Perea-Romero, I.; Swafiri, S.T.; Zurita, O.; et al. Comprehensive Genotyping and Phenotyping Analysis of GUCY2D-Associated Rod- and Cone-Dominated Dystrophies. Am. J. Ophthalmol. 2023, 254, 87–103. [Google Scholar] [CrossRef]
- Sun, Z.; Wu, S.; Zhu, T.; Li, H.; Wei, X.; Du, H.; Sui, R. Variants at codon 838 in the GUCY2D gene result in different phenotypes of cone rod dystrophy. Ophthalmic Genet. 2020, 41, 548–555. [Google Scholar] [CrossRef]
- Xiao, X.; Guo, X.; Jia, X.; Li, S.; Wang, P.; Zhang, Q. A recurrent mutation in GUCY2D associated with autosomal dominant cone dystrophy in a Chinese family. Mol. Vis. 2011, 17, 3271–3278. [Google Scholar]
- Zobor, D.; Zrenner, E.; Wissinger, B.; Kohl, S.; Jagle, H. GUCY2D- or GUCA1A-related autosomal dominant cone-rod dystrophy: Is there a phenotypic difference? Retina 2014, 34, 1576–1587. [Google Scholar] [CrossRef]
- Lazar, C.H.; Mutsuddi, M.; Kimchi, A.; Zelinger, L.; Mizrahi-Meissonnier, L.; Marks-Ohana, D.; Boleda, A.; Ratnapriya, R.; Sharon, D.; Swaroop, A.; et al. Whole exome sequencing reveals GUCY2D as a major gene associated with cone and cone-rod dystrophy in Israel. Investig. Ophthalmol. Vis. Sci. 2014, 56, 420–430. [Google Scholar] [CrossRef]
- Udar, N.; Yelchits, S.; Chalukya, M.; Yellore, V.; Nusinowitz, S.; Silva-Garcia, R.; Vrabec, T.; Hussles Maumenee, I.; Donoso, L.; Small, K.W. Identification of GUCY2D gene mutations in CORD5 families and evidence of incomplete penetrance. Hum. Mutat. 2003, 21, 170–171. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Wimberg, H.; Kinarty, Y.; Koch, K.W. Genotype-functional-phenotype correlations in photoreceptor guanylate cyclase (GC-E) encoded by GUCY2D. Prog. Retin. Eye Res. 2018, 63, 69–91. [Google Scholar] [CrossRef] [PubMed]
- Daich Varela, M.; Georgiadis, A.; Michaelides, M. Genetic treatment for autosomal dominant inherited retinal dystrophies: Approaches, challenges and targeted genotypes. Br. J. Ophthalmol. 2023, 107, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N.; Georgiou, M.; Khan, K.N.; Michaelides, M. Macular dystrophies: Clinical and imaging features, molecular genetics and therapeutic options. Br. J. Ophthalmol. 2020, 104, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Michaelides, M.; Hunt, D.M.; Moore, A.T. The genetics of inherited macular dystrophies. J. Med. Genet. 2003, 40, 641–650. [Google Scholar] [CrossRef]
- Kumaran, N.; Pennesi, M.E.; Yang, P.; Trzupek, K.M.; Schlechter, C.; Moore, A.T.; Weleber, R.G.; Michaelides, M. Leber Congenital Amaurosis/Early-Onset Severe Retinal Dystrophy Overview. In GeneReviews((R)); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2023. [Google Scholar]
- Daich Varela, M.; Cabral de Guimaraes, T.A.; Georgiou, M.; Michaelides, M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Current management and clinical trials. Br. J. Ophthalmol. 2022, 106, 445–451. [Google Scholar] [CrossRef]
- Kumaran, N.; Moore, A.T.; Weleber, R.G.; Michaelides, M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Clinical features, molecular genetics and therapeutic interventions. Br. J. Ophthalmol. 2017, 101, 1147–1154. [Google Scholar] [CrossRef]
- Hunt, D.M.; Buch, P.; Michaelides, M. Guanylate cyclases and associated activator proteins in retinal disease. Mol. Cell Biochem. 2010, 334, 157–168. [Google Scholar] [CrossRef]
- Michaelides, M.; Hardcastle, A.J.; Hunt, D.M.; Moore, A.T. Progressive cone and cone-rod dystrophies: Phenotypes and underlying molecular genetic basis. Surv. Ophthalmol. 2006, 51, 232–258. [Google Scholar] [CrossRef]
- Robson, A.G.; Michaelides, M.; Saihan, Z.; Bird, A.C.; Webster, A.R.; Moore, A.T.; Fitzke, F.W.; Holder, G.E. Functional characteristics of patients with retinal dystrophy that manifest abnormal parafoveal annuli of high density fundus autofluorescence; a review and update. Doc. Ophthalmol. 2008, 116, 79–89. [Google Scholar] [CrossRef]
- Gill, J.S.; Georgiou, M.; Kalitzeos, A.; Moore, A.T.; Michaelides, M. Progressive cone and cone-rod dystrophies: Clinical features, molecular genetics and prospects for therapy. Br. J. Ophthalmol. 2019, 103, 711–720. [Google Scholar] [CrossRef]
- Thompson, J.A.; De Roach, J.N.; McLaren, T.L.; Montgomery, H.E.; Hoffmann, L.H.; Campbell, I.R.; Chen, F.K.; Mackey, D.A.; Lamey, T.M. The genetic profile of Leber congenital amaurosis in an Australian cohort. Mol. Genet. Genom. Med. 2017, 5, 652–667. [Google Scholar] [CrossRef]
- Huang, L.; Xiao, X.; Li, S.; Jia, X.; Wang, P.; Guo, X.; Zhang, Q. CRX variants in cone-rod dystrophy and mutation overview. Biochem. Biophys. Res. Commun. 2012, 426, 498–503. [Google Scholar] [CrossRef]
- Sakuramoto, H.; Kuniyoshi, K.; Tsunoda, K.; Akahori, M.; Iwata, T.; Shimomura, Y. Two siblings with late-onset cone-rod dystrophy and no visible macular degeneration. Clin. Ophthalmol. 2013, 7, 1703–1711. [Google Scholar] [CrossRef]
- Zhu, L.; Ouyang, W.; Zhang, M.; Wang, H.; Li, S.; Meng, X.; Yin, Z.Q. Molecular genetics with clinical characteristics of Leber congenital amaurosis in the Han population of western China. Ophthalmic Genet. 2021, 42, 392–401. [Google Scholar] [CrossRef]
- Chen, S.; Peng, G.H.; Wang, X.; Smith, A.C.; Grote, S.K.; Sopher, B.L.; La Spada, A.R. Interference of Crx-dependent transcription by ataxin-7 involves interaction between the glutamine regions and requires the ataxin-7 carboxy-terminal region for nuclear localization. Hum. Mol. Genet. 2004, 13, 53–67. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Q.L.; Nie, Z.; Sun, H.; Lennon, G.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Zack, D.J. Crx, a novel Otx-like paired-homeodomain protein, binds to and transactivates photoreceptor cell-specific genes. Neuron 1997, 19, 1017–1030. [Google Scholar] [CrossRef]
- Furukawa, T.; Morrow, E.M.; Cepko, C.L. Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell 1997, 91, 531–541. [Google Scholar] [CrossRef]
- Itabashi, T.; Wada, Y.; Sato, H.; Kunikata, H.; Kawamura, M.; Tamai, M. Ocular findings in a Japanese family with an Arg41Trp mutation of the CRX gene. Graefes Arch. Clin. Exp. Ophthalmol. 2003, 241, 535–540. [Google Scholar] [CrossRef]
- Kimura, A.; Singh, D.; Wawrousek, E.F.; Kikuchi, M.; Nakamura, M.; Shinohara, T. Both PCE-1/RX and OTX/CRX interactions are necessary for photoreceptor-specific gene expression. J. Biol. Chem. 2000, 275, 1152–1160. [Google Scholar] [CrossRef]
- Peng, G.H.; Ahmad, O.; Ahmad, F.; Liu, J.; Chen, S. The photoreceptor-specific nuclear receptor Nr2e3 interacts with Crx and exerts opposing effects on the transcription of rod versus cone genes. Hum. Mol. Genet. 2005, 14, 747–764. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, S.; Guo, X.; Guo, L.; Xiao, X.; Jia, X.; Kuang, Z. Screening for CRX gene mutations in Chinese patients with Leber congenital amaurosis and mutational phenotype. Ophthalmic Genet. 2001, 22, 89–96. [Google Scholar] [CrossRef]
- Perrault, I.; Rozet, J.M.; Calvas, P.; Gerber, S.; Camuzat, A.; Dollfus, H.; Chatelin, S.; Souied, E.; Ghazi, I.; Leowski, C.; et al. Retinal-specific guanylate cyclase gene mutations in Leber’s congenital amaurosis. Nat. Genet. 1996, 14, 461–464. [Google Scholar] [CrossRef]
- Freund, C.L.; Gregory-Evans, C.Y.; Furukawa, T.; Papaioannou, M.; Looser, J.; Ploder, L.; Bellingham, J.; Ng, D.; Herbrick, J.A.; Duncan, A.; et al. Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell 1997, 91, 543–553. [Google Scholar] [CrossRef]
- Akagi, T.; Mandai, M.; Ooto, S.; Hirami, Y.; Osakada, F.; Kageyama, R.; Yoshimura, N.; Takahashi, M. Otx2 homeobox gene induces photoreceptor-specific phenotypes in cells derived from adult iris and ciliary tissue. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4570–4575. [Google Scholar] [CrossRef]
- Hull, S.; Arno, G.; Plagnol, V.; Chamney, S.; Russell-Eggitt, I.; Thompson, D.; Ramsden, S.C.; Black, G.C.; Robson, A.; Holder, G.E.; et al. The phenotypic variability of retinal dystrophies associated with mutations in CRX, with report of a novel macular dystrophy phenotype. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6934–6944. [Google Scholar] [CrossRef]
- Al-Khuzaei, S.; Hudspith, K.A.Z.; Broadgate, S.; Shanks, M.E.; Clouston, P.; Nemeth, A.H.; Halford, S.; Downes, S.M. Targeted next generation sequencing and family survey enable correct genetic diagnosis in CRX associated macular dystrophy—A case report. BMC Ophthalmol. 2021, 21, 168. [Google Scholar] [CrossRef]
- D’Esposito, F.; Cennamo, G.; de Crecchio, G.; Maltese, P.E.; Cecchin, S.; Bertelli, M.; Ziccardi, L.; Esposito Veneruso, P.; Magli, A.; Cennamo, G.; et al. Multimodal Imaging in Autosomal Dominant Cone-Rod Dystrophy Caused by Novel CRX Variant. Ophthalmic Res. 2018, 60, 169–175. [Google Scholar] [CrossRef]
- Griffith, J.F.; DeBenedictis, M.J.; Traboulsi, E.I. A novel dominant CRX mutation causes adult-onset macular dystrophy. Ophthalmic Genet. 2018, 39, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.T.; Alarcon-Martinez, T.; Lopez, I.; Fajardo, N.; Chiang, J.; Koenekoop, R.K. A complete, homozygous CRX deletion causing nullizygosity is a new genetic mechanism for Leber congenital amaurosis. Sci. Rep. 2018, 8, 5034. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.G.; Joo, K.; Han, J.; Choi, M.; Kim, S.W.; Park, K.H.; Park, S.J.; Lee, C.S.; Byeon, S.H.; Woo, S.J. Genotypic Profile and Clinical Characteristics of CRX-Associated Retinopathy in Koreans. Genes 2023, 14, 1057. [Google Scholar] [CrossRef]
- Lines, M.A.; Hebert, M.; McTaggart, K.E.; Flynn, S.J.; Tennant, M.T.; MacDonald, I.M. Electrophysiologic and phenotypic features of an autosomal cone-rod dystrophy caused by a novel CRX mutation. Ophthalmology 2002, 109, 1862–1870. [Google Scholar] [CrossRef]
- Nasser, F.; Kurtenbach, A.; Kohl, S.; Obermaier, C.; Stingl, K.; Zrenner, E. Retinal dystrophies with bull’s-eye maculopathy along with negative ERGs. Doc. Ophthalmol. 2019, 139, 45–57. [Google Scholar] [CrossRef]
- Nishiguchi, K.M.; Kunikata, H.; Fujita, K.; Hashimoto, K.; Koyanagi, Y.; Akiyama, M.; Ikeda, Y.; Momozawa, Y.; Sonoda, K.H.; Murakami, A.; et al. Association of CRX genotypes and retinal phenotypes confounded by variable expressivity and electronegative electroretinogram. Clin. Exp. Ophthalmol. 2020, 48, 644–657. [Google Scholar] [CrossRef]
- Paunescu, K.; Preising, M.N.; Janke, B.; Wissinger, B.; Lorenz, B. Genotype-phenotype correlation in a German family with a novel complex CRX mutation extending the open reading frame. Ophthalmology 2007, 114, 1348–1357. [Google Scholar] [CrossRef]
- Romdhane, K.; Vaclavik, V.; Schorderet, D.F.; Munier, F.L.; Viet Tran, H. CRX-linked macular dystrophy with intrafamilial variable expressivity. Ophthalmic Genet. 2018, 39, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Swaroop, A.; Wang, Q.L.; Wu, W.; Cook, J.; Coats, C.; Xu, S.; Chen, S.; Zack, D.J.; Sieving, P.A. Leber congenital amaurosis caused by a homozygous mutation (R90W) in the homeodomain of the retinal transcription factor CRX: Direct evidence for the involvement of CRX in the development of photoreceptor function. Hum. Mol. Genet. 1999, 8, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Yahya, S.; Smith, C.E.L.; Poulter, J.A.; McKibbin, M.; Arno, G.; Ellingford, J.; Kampjarvi, K.; Khan, M.I.; Cremers, F.P.M.; Hardcastle, A.J.; et al. Late-Onset Autosomal Dominant Macular Degeneration Caused by Deletion of the CRX Gene. Ophthalmology 2023, 130, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Xiao, X.; Li, S.; Sun, W.; Zhang, Q. Pathogenicity discrimination and genetic test reference for CRX variants based on genotype-phenotype analysis. Exp. Eye Res. 2019, 189, 107846. [Google Scholar] [CrossRef]
- Freund, C.L.; Wang, Q.L.; Chen, S.; Muskat, B.L.; Wiles, C.D.; Sheffield, V.C.; Jacobson, S.G.; McInnes, R.R.; Zack, D.J.; Stone, E.M. De novo mutations in the CRX homeobox gene associated with Leber congenital amaurosis. Nat. Genet. 1998, 18, 311–312. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RP1L1 | CRX | GUCY2D | Other OMD | ||
|---|---|---|---|---|---|
| Inheritance (number of families) | AD | 23 | 1 | 1 | 5 |
| AR | 0 | 0 | 0 | 2 | |
| Sporadic | 7 | 0 | 1 | 10 | |
| Age (years) † | 49 (6–88) | 54 (40–68) | 47.5 (40–55) | 55 (15–91) | |
| Age at onset (years) † | 30 (2–71) | 49.5 (31–68) | 26.5 (13–40) | 45 (6–49) | |
| Disease duration (years) † | 11 (0–63) | 4.5 (0–9) | 21 (0–42) | 8 (0–51) | |
| BCVA (logMAR) † | Right eye | 0.52 (−0.08–1.52) | 0.07 (−0.08–0.22) | 0.91 (0.82–1) | 0.4 (0.18–1.4) |
| Light eye | 0.52 (−0.08–1.7) | 0.16 (−0.08–0.4) | 0.96 (0.82–1.1) | 0.4 (0.18–1.4) | |
| Visual field pattern †† | Pattern 1 | 32 | 0 | 2 | 16 |
| Pattern 2 | 11 | 2 | 0 | 4 | |
| mfERG group § | Group 1 | 3 | 2 | 0 | 4 |
| Group 2 | 28 | 0 | 2 | 12 | |
| Group 3 | 6 | 0 | 0 | 1 | |
| SD-OCT classification §§ | Classical | 41 | 0 | 0 | 1 |
| Non-classical | 6 | 2 | 2 | 20 | |
| Gene | Nucleotide Change, Amino Acid Change | State | Family Number | Reference |
|---|---|---|---|---|
| RP1L1 | c.133C>T, p.Arg45Trp | Het | 16 | Akahori et al. (2010) [33], Tsunoda et al. (2012) [25], Fujinami et al. (2019) [6], Wang et al. (2020) [15] |
| RP1L1 | c.3581C>T, p.Thr1194Met/c.3587C>T, p.Thr1196Ile | Het | 1 | Fujinami et al. (2016) [11] |
| RP1L1 | c.3593C>T, p.Ser1198Phe | Het | 1 | Fujinami et al. (2019) [6] |
| RP1L1 | c.3595T>C, p.Ser1199Pro | Het | 1 | Takahashi H et al. (2014) [31], Fujinami et al. (2019) [6] |
| RP1L1 | c.3596C>G, p.Ser1199Cys | Het | 7 | Kabuto et al. (2012) [32], Fujinami et al. (2019) [6] |
| RP1L1 | c.3599G>A, p.Gly1200Asp | Het | 1 | Fujinami et al. (2016) [11] |
| RP1L1 | c.3599G>C, p.Gly1200Ala | Het | 1 | Fujinami et al. (2019) [6] |
| RP1L1 | c.3602T>G, p.Val1201Gly | Het | 1 | Fujinami et al. (2016) [11] |
| RP1L1 | c.3602T>C, p.Val1201Ala | Het | 1 | This study |
| CRX | c.128G>A, p.Arg43His | Het | 1 | Fujinami-Yokokawa et al. (2020) [39] |
| GUCY2D | c.2747T>C, p.Ile916Thr | Het | 1 | de Castro-Miró et al. (2014) [49], Liu et al. (2020) [40] |
| GUCY2D | c.2513G>A, p.Arg838His | Het | 1 | Payne et al. (2001) [48], Liu et al. (2020) [40] |
| Gene | Variant | Previous Report | Inheritance | Phenotype (Family Number) | Presence with Normal Fundus (Subject Number) |
|---|---|---|---|---|---|
| CRX | c.128G>A, p.Arg43His | Fujinami-Yokokawa et al. (2020) [59] | AD | OMD (1) | Yes (2) |
| GUCY2D | c.2747T>C, p.Ile916Thr | de Castro-Miró et al. (2014) [49] | AD | COD (1) | NA |
| Liu et al. (2020) [40] | AD | OMD (1) | Yes (1) | ||
| Rodilla C et al. (2023) [56] | AD | CORD (1) | NA | ||
| c.2513G>A, p.Arg838His | Payne et al. (2001) [48] | AD | CORD (1) | No(1) | |
| Ito et al. (2004) [50] | AD | COD (1) | Yes (1), no (1) | ||
| Weigell-Weber et al. (2000) [52] | AD | CORD (1) | NA | ||
| Lazar et al. (2015) [60] | AD | COD (2), CORD (1) | No (2) | ||
| Kim et al. (2019) [16] | AD | COD (1) | NA | ||
| Sharon et al. (2019) [62] | AD | COD (1) | NA | ||
| Udar et al. (2003) [61] | AD | CORD (1) | NA | ||
| Kitiratschky et al. (2008) [54] | AD | COD (3) | Yes (1), no (6) | ||
| Xiao et al. (2011) [58] | AD | COD (1) | No (8) | ||
| Mukherjee et al. (2014) [55] | AD (de novo) | COD (1) | No (3) | ||
| Zobor et al. (2014) [59] | AD | COD/CORD (1) | Yes (1), no (2) | ||
| Jiang et al. (2015) [53] | AD | COD (4), CORD (1) | No (6) | ||
| Sun et al. (2020) [57] | AD | CORD (1) | No (2) | ||
| Rodilla C et al. (2023) [56] | AD | CORD (8) | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujinami-Yokokawa, Y.; Yang, L.; Joo, K.; Tsunoda, K.; Liu, X.; Kondo, M.; Ahn, S.J.; Li, H.; Park, K.H.; Tachimori, H.; et al. Occult Macular Dysfunction Syndrome: Identification of Multiple Pathologies in a Clinical Spectrum of Macular Dysfunction with Normal Fundus in East Asian Patients: EAOMD Report No. 5. Genes 2023, 14, 1869. https://doi.org/10.3390/genes14101869
Fujinami-Yokokawa Y, Yang L, Joo K, Tsunoda K, Liu X, Kondo M, Ahn SJ, Li H, Park KH, Tachimori H, et al. Occult Macular Dysfunction Syndrome: Identification of Multiple Pathologies in a Clinical Spectrum of Macular Dysfunction with Normal Fundus in East Asian Patients: EAOMD Report No. 5. Genes. 2023; 14(10):1869. https://doi.org/10.3390/genes14101869
Chicago/Turabian StyleFujinami-Yokokawa, Yu, Lizhu Yang, Kwangsic Joo, Kazushige Tsunoda, Xiao Liu, Mineo Kondo, Seong Joon Ahn, Hui Li, Kyu Hyung Park, Hisateru Tachimori, and et al. 2023. "Occult Macular Dysfunction Syndrome: Identification of Multiple Pathologies in a Clinical Spectrum of Macular Dysfunction with Normal Fundus in East Asian Patients: EAOMD Report No. 5" Genes 14, no. 10: 1869. https://doi.org/10.3390/genes14101869