

Drosophila melanogaster as a Model to Study Fragile X-Associated Disorders
 , and
, and 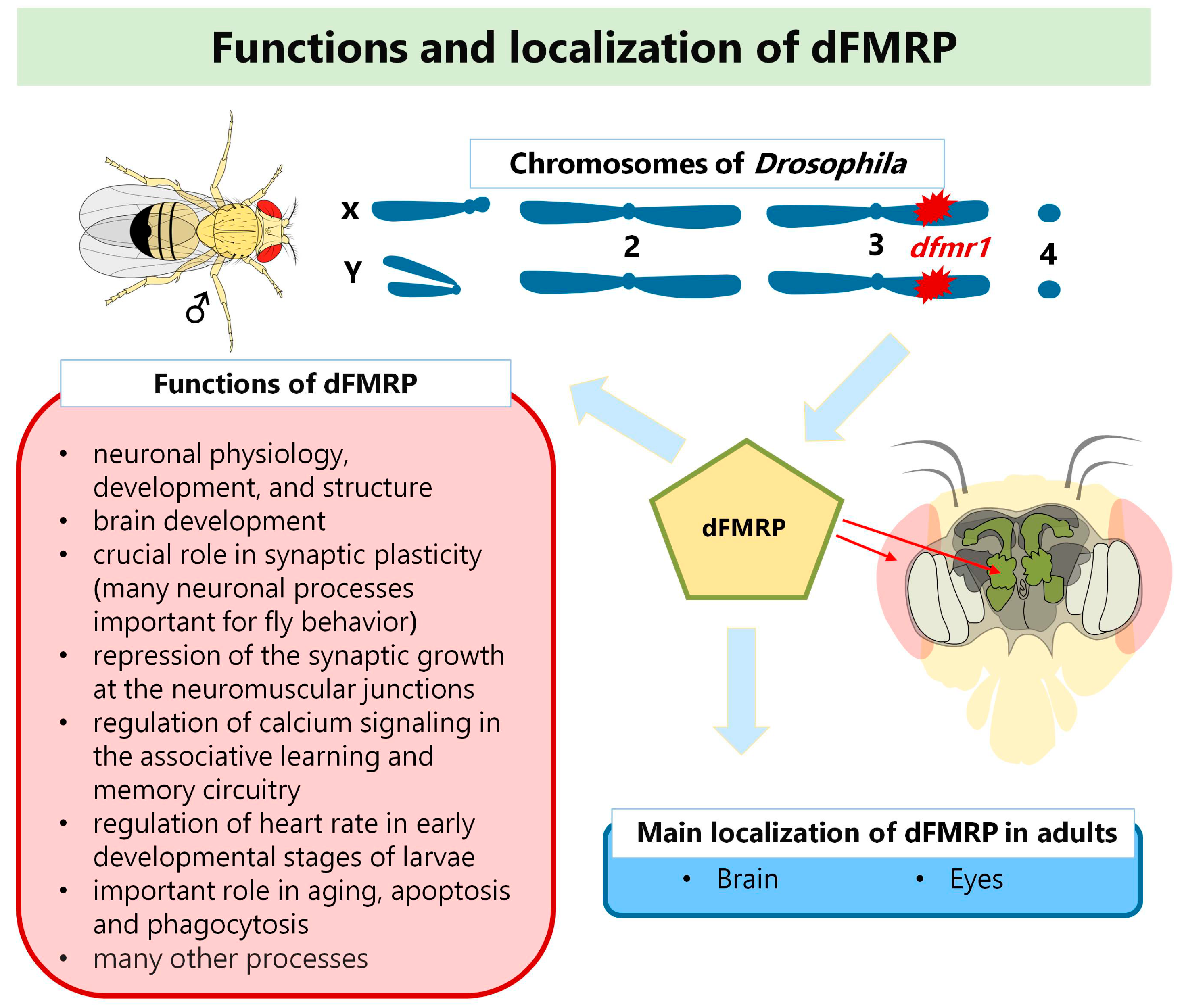{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Fragile X Syndrome
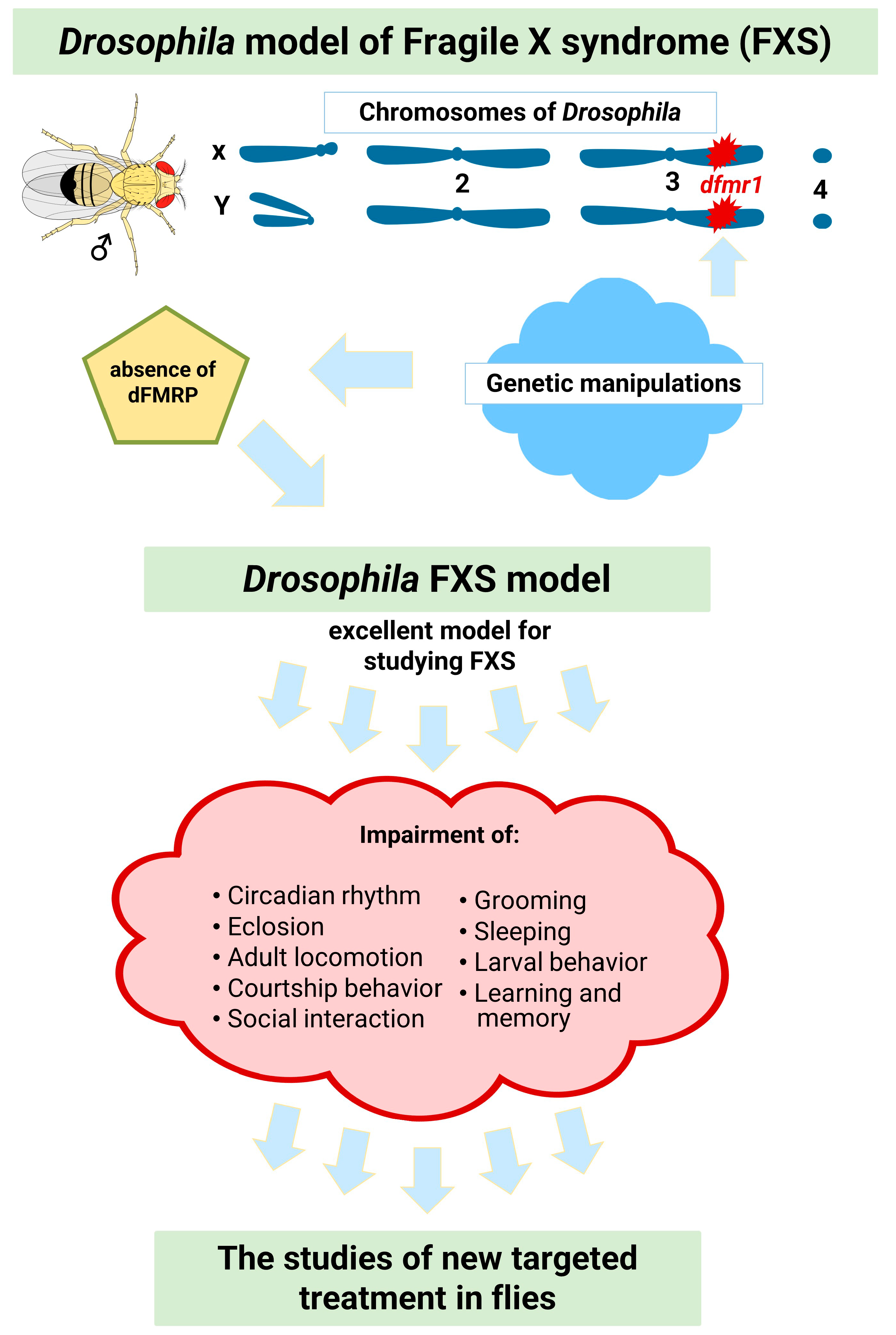
3. Drosophila Model for the Study of Fragile X Syndrome
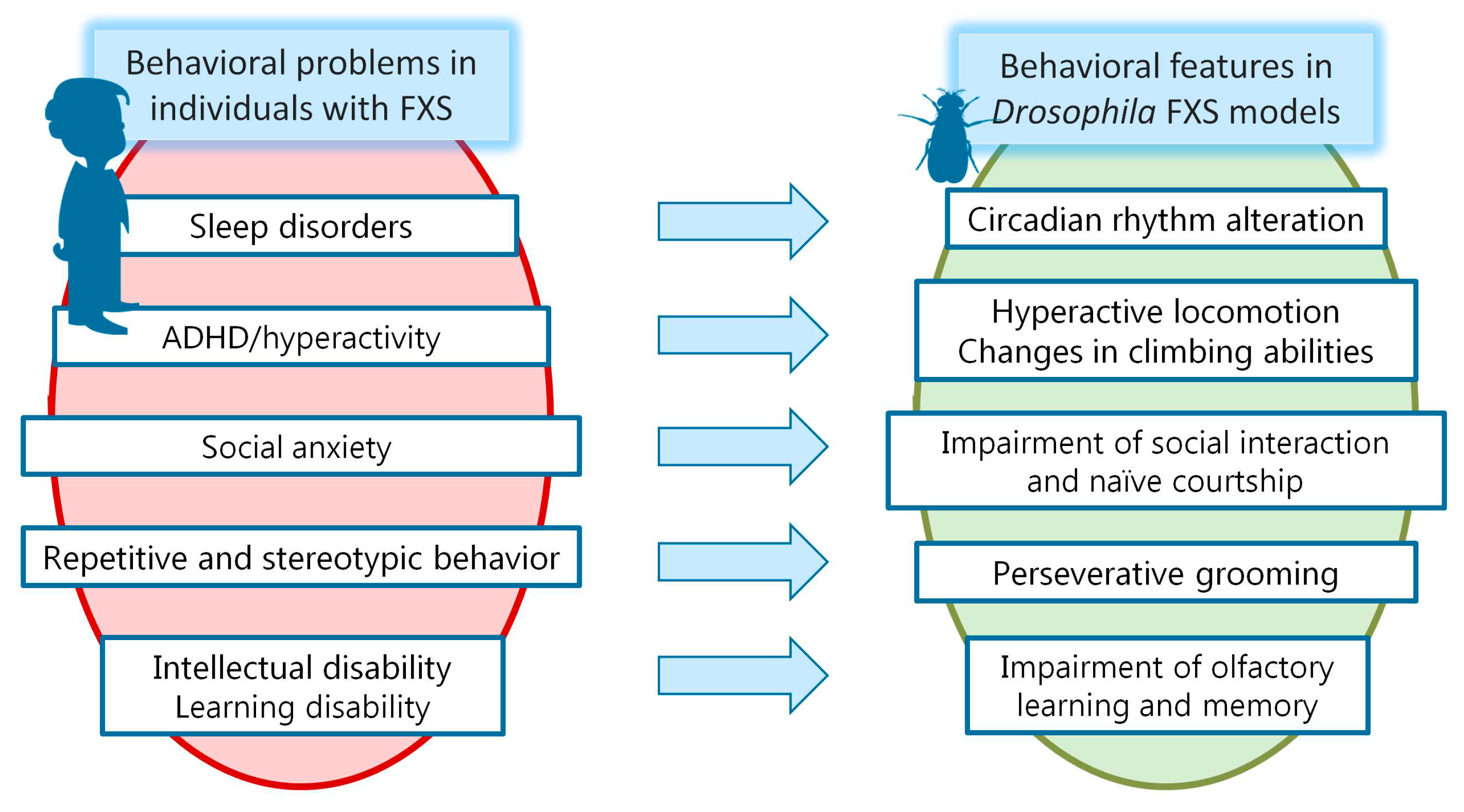
3.1. Impaired Circadian Rhythms and Sleep Problems in FXS
3.2. Hyperactivity and Attention Deficit/Hyperactivity Disorder in FXS
3.3. Other Autistic-Like Behaviors in FXS
3.4. ID in FXS and Learning and Memory Impairment in the FXS Model of Drosophila
4. D. melanogaster as a Model Organism to Study Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS)
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- King, R.C. Genetics, 2nd ed.; Oxford University Press: New York, NY, USA, 1965; pp. 1–462. [Google Scholar]
- Fuentes, E.; Sivakumar, N.; Selvik, L.-K.; Arch, M.; Cardona, P.J.; Ioerger, T.R.; Dragset, M.S. Drosophila melanogaster is a powerful host model to study mycobacterial virulence. bioRxiv 2022. [Google Scholar] [CrossRef]
- Russell, W.M.S.; Burch, R.L. The Principles of Humane Experimental Technique; Methuen & Co, Ltd.: North Yorkshire, UK, 1959; p. 238. [Google Scholar]
- Adams, M.D.; Celniker, S.E.; Holt, R.A.; Evans, C.A.; Gocayne, J.D.; Amanatides, P.G.; Scherer, S.E.; Li, P.W.; Hoskins, R.A.; Galle, R.F.; et al. The genome sequence of Drosophila melanogaster. Science 2000, 287, 2185–2195. [Google Scholar] [CrossRef] [Green Version]
- Myers, E.W.; Sutton, G.G.; Delcher, A.L.; Dew, I.M.; Fasulo, D.P.; Flanigan, M.J.; Kravitz, S.A.; Mobarry, C.M.; Reinert, K.H.; Remington, K.A.; et al. A whole-genome assembly of Drosophila. Science 2000, 287, 2196–2204. [Google Scholar] [CrossRef] [PubMed]
- Celniker, S.E.; Wheeler, D.A.; Kronmiller, B.; Carlson, J.W.; Halpern, A.; Patel, S.; Adams, M.; Champe, M.; Dugan, S.P.; Frise, E.; et al. Finishing a whole-genome shotgun: Release 3 of the Drosophila melanogaster euchromatic genome sequence. Genome Biol. 2002, 3, research0079.1. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.B.; Boley, N.; Eisman, R.; May, G.E.; Stoiber, M.H.; Duff, M.O.; Booth, B.W.; Wen, J.; Park, S.; Suzuki, A.M.; et al. Diversity and dynamics of the Drosophila transcriptome. Nature 2014, 512, 393–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, T.C. A Short History and Description of Drosophila melanogaster Classical Genetics: Chromosome Aberrations, Forward Genetic Screens, and the Nature of Mutations. Genetics 2017, 206, 665–689. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Jaiswal, M.; Charng, W.-L.; Gambin, T.; Karaca, E.; Mirzaa, G.; Wiszniewski, W.; Sandoval, H.; Haelterman, N.A.; Xiong, B.; et al. A Drosophila Genetic Resource of Mutants to Study Mechanisms Underlying Human Genetic Diseases. Cell 2014, 159, 200–214. [Google Scholar] [CrossRef] [Green Version]
- Ugur, B.; Chen, K.; Bellen, H.J. Drosophila tools and assays for the study of human diseases. Dis. Model. Mech. 2016, 9, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Gehring, W.J.; Kloter, U.; Suga, H. Chapter 2 Evolution of the Hox Gene Complex from an Evolutionary Ground State. In Current Topics in Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2009; Volume 88, pp. 35–61. [Google Scholar]
- Jennings, B.H. Drosophila–a versatile model in biology & medicine. Mater. Today 2011, 14, 190–195. [Google Scholar]
- O’Kane, C.J. Drosophila as a model organism for the study of neuropsychiatric disorders. Curr. Top Behav. Neurosci. 2011, 7, 37–60. [Google Scholar]
- Nitta, Y.; Sugie, A. Studies of neurodegenerative diseases using Drosophila and the development of novel approaches for their analysis. Fly 2022, 16, 275–298. [Google Scholar] [CrossRef]
- Campbell, R.A.A.; Turner, G.C. The mushroom body. Curr. Biol. 2010, 20, R11–R12. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Yoshida, H. Drosophila as a Model Organism. In Drosophila Models for Human Diseases; Advances in Experimental Medicine and, Biology; Yamaguchi, M., Ed.; Springer: Singapore, 2018; Volume 1076, pp. 1–10. [Google Scholar]
- Bellen, H.J.; Tong, C.; Tsuda, H. 100 years of Drosophila research and its impact on vertebrate neuroscience: A history lesson for the future. Nat. Rev. Neurosci. 2010, 11, 514–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortini, M.E.; Bonini, N.M. Modeling human neurodegenerative diseases in Drosophila: On a wing and a prayer. Trends Genet. 2000, 16, 161–167. [Google Scholar] [CrossRef]
- Muqit, M.M.K.; Feany, M.B. Modelling neurodegenerative diseases in Drosophila: A fruitful approach? Nat. Rev. Neurosci. 2002, 3, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Verheyen, E.M. The power of Drosophila in modeling human disease mechanisms. Dis. Model. Mech. 2022, 15, dmm049549. [Google Scholar] [CrossRef]
- Drozd, M.; Bardoni, B.; Capovilla, M. Modeling Fragile X Syndrome in Drosophila. Front. Mol. Neurosci. 2018, 11, 124. [Google Scholar] [CrossRef] [PubMed]
- Curnow, E.; Wang, Y. New Animal Models for Understanding FMRP Functions and FXS Pathology. Cells 2022, 11, 1628. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Weiler, I.J.; Irwin, S.A.; Klintsova, A.Y.; Spencer, C.M.; Brazelton, A.D.; Miyashiro, K.; Comery, T.A.; Patel, B.; Eberwine, J.; Greenough, W.T. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc. Natl. Acad. Sci. USA 1997, 94, 5395–5400. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Gutekunst, C.A.; Eberhart, D.E.; Yi, H.; Warren, S.T.; Hersch, S.M. Fragile X mental retardation protein: Nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J. Neurosci. 1997, 17, 1539–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagerman, R.J.; Hagerman, P.J. Testing for fragile X gene mutations throughout the life span. JAMA 2008, 300, 2419–2421. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, J.S.; Nelson, D.L.; Zhang, F.; Pieretti, M.; Caskey, C.T.; Saxe, D.; Warren, S.T. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1992, 1, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Saldarriaga, W.; Tassone, F.; González-Teshima, L.Y.; Forero-Forero, J.V.; Ayala-Zapata, S.; Hagerman, R. Fragile X Syndrome. Colomb. Med. 2014, 45, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Tassone, F.; Iong, K.P.; Tong, T.H.; Lo, J.; Gane, L.W.; Berry-Kravis, E.; Nguyen, D.; Mu, L.Y.; Laffin, J.; Bailey, D.B.; et al. FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States. Genome Med. 2012, 4, 100. [Google Scholar] [CrossRef] [Green Version]
- Devys, D.; Lutz, Y.; Rouyer, N.; Bellocq, J.P.; Mandel, J.L. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat. Genet. 1993, 4, 335–340. [Google Scholar] [CrossRef]
- Bardoni, B.; Mandel, J.L.; Fisch, G.S. FMR1 gene and fragile X syndrome. Am. J. Med. Genet. 2000, 97, 153–163. [Google Scholar] [CrossRef]
- Hoogeveen, A.T.; Willemsen, R.; Oostra, B.A. Fragile X syndrome, the Fragile X related proteins, and animal models. Microsc. Res. Tech. 2002, 57, 148–155. [Google Scholar] [CrossRef]
- Bardoni, B.; Davidovic, L.; Bensaid, M.; Khandjian, E.W. The fragile X syndrome: Exploring its molecular basis and seeking a treatment. Expert Rev. Mol. Med. 2006, 8, 1–16. [Google Scholar] [CrossRef]
- Maurin, T.; Zongaro, S.; Bardoni, B. Fragile X Syndrome: From molecular pathology to therapy. Neurosci. Biobehav. Rev. 2014, 46 Pt 2, 242–255. [Google Scholar] [CrossRef]
- Davidovic, L.; Jaglin, X.H.; Lepagnol-Bestel, A.M.; Tremblay, S.; Simonneau, M.; Bardoni, B.; Khandjian, E.W. The fragile X mental retardation protein is a molecular adaptor between the neurospecific KIF3C kinesin and dendritic RNA granules. Hum Mol. Genet. 2007, 16, 3047–3058. [Google Scholar] [CrossRef] [PubMed]
- Kelley, K.; Chang, S.J.; Lin, S.L. Mechanism of repeat-associated microRNAs in fragile X syndrome. Neural Plast. 2012, 2012, 104796. [Google Scholar] [CrossRef] [PubMed]
- Specchia, V.; D’Attis, S.; Puricella, A.; Bozzetti, M.P. dFmr1 Plays Roles in Small RNA Pathways of Drosophila melanogaster. Int. J. Mol. Sci. 2017, 18, 1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budimirovic, D.B.; Subramanian, M. Neurobiology of Autism and Intellectual Disability: Fragile X Syndrome. In Neurobiology of Disease, 2nd ed.; Johnston, M., Michael, A.M., Jr., Fatemi, M.H., Ali, M.B.A., Eds.; Oxford University Press: New York, NY, USA, 2016; pp. 375–384. [Google Scholar]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B., Jr.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.B., Jr.; Raspa, M.; Olmsted, M.; Holiday, D.B. Co-occurring conditions associated with FMR1 gene variations: Findings from a national parent survey. Am. J. Med. Genet. A 2008, 146A, 2060–2069. [Google Scholar] [CrossRef]
- Kaufmann, W.E.; Kidd, S.A.; Andrews, H.F.; Budimirovic, D.B.; Esler, A.; Haas-Givler, B.; Stackhouse, T.; Riley, C.; Peacock, G.; Sherman, S.L.; et al. Autism Spectrum Disorder in Fragile X Syndrome: Cooccurring Conditions and Current Treatment. Pediatrics 2017, 139 (Suppl. S3), S194–S206. [Google Scholar] [CrossRef] [Green Version]
- Budimirovic, D.; Haas-Givler, B.; Blitz, R.; Esler, A.; Kaufmann, W.; Sudhalter, V.; Stackhouse, T.M.; Scharfenaker, S.K.; Berry-Kravis, E. Autism Spectrum Disorder in Fragile X Syndrome. Available online: https://fragilex.org/wp-content/uploads/2012/08/Autism-Spectrum-Disorder-in-Fragile-X-Syndrome-2014-Nov.pdf (accessed on 5 October 2022).
- Budimirovic, D.B.; Protic, D.D.; Delahunty, C.M.; Andrews, H.F.; Choo, T.H.; Xu, Q.; Berry-Kravis, E.; Kaufmann, W.E.; Consortium, F. Sleep problems in fragile X syndrome: Cross-sectional analysis of a large clinic-based cohort. Am. J. Med. Genet. A 2022, 188, 1029–1039. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Filipink, R.A.; Frye, R.E.; Golla, S.; Morris, S.M.; Andrews, H.; Choo, T.H.; Kaufmann, W.E.; Consortium, F. Seizures in Fragile X Syndrome: Associations and Longitudinal Analysis of a Large Clinic-Based Cohort. Front. Pediatr. 2021, 9, 736255. [Google Scholar] [CrossRef]
- Choo, T.H.; Xu, Q.; Budimirovic, D.; Lozano, R.; Esler, A.N.; Frye, R.E.; Andrews, H.; Velinov, M. Height and BMI in fragile X syndrome: A longitudinal assessment. Obesity 2022, 30, 743–750. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Hagerman, P.J. Fragile X Syndrome and Premutation Disorders; Mac Keith Press: London, UK, 2020. [Google Scholar]
- Dahlhaus, R. Of Men and Mice: Modeling the Fragile X Syndrome. Front. Mol. Neurosci. 2018, 11, 41. [Google Scholar] [CrossRef]
- Wan, L.; Dockendorff, T.C.; Jongens, T.A.; Dreyfuss, G. Characterization of dFMR1, a Drosophila melanogaster homolog of the fragile X mental retardation protein. Mol. Cell Biol. 2000, 20, 8536–8547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.Q.; Bailey, A.M.; Matthies, H.J.; Renden, R.B.; Smith, M.A.; Speese, S.D.; Rubin, G.M.; Broadie, K. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 2001, 107, 591–603. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.; Hiesinger, P.R.; Schroeder, A.J.; Kume, K.; Verstreken, P.; Jackson, F.R.; Nelson, D.L.; Hassan, B.A. Drosophila Fragile X Protein, DFXR, Regulates Neuronal Morphology and Function in the Brain. Neuron 2002, 34, 961–972. [Google Scholar] [CrossRef] [Green Version]
- Sudhakaran, I.P.; Hillebrand, J.; Dervan, A.; Das, S.; Holohan, E.E.; Hulsmeier, J.; Sarov, M.; Parker, R.; VijayRaghavan, K.; Ramaswami, M. FMRP and Ataxin-2 function together in long-term olfactory habituation and neuronal translational control. Proc. Natl. Acad. Sci. USA 2014, 111, E99–E108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, L.; Zhang, Y.Q.; Woodruff, E.; Broadie, K. The Drosophila fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr. Biol. 2004, 14, 1863–1870. [Google Scholar] [CrossRef] [Green Version]
- Inoue, S.; Shimoda, M.; Nishinokubi, I.; Siomi, M.C.; Okamura, M.; Nakamura, A.; Kobayashi, S.; Ishida, N.; Siomi, H. A role for the Drosophila fragile X-related gene in circadian output. Curr. Biol. 2002, 12, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Dockendorff, T.C.; Su, H.S.; McBride, S.M.; Yang, Z.; Choi, C.H.; Siwicki, K.K.; Sehgal, A.; Jongens, T.A. Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron 2002, 34, 973–984. [Google Scholar] [CrossRef] [Green Version]
- McBride, S.M.; Holloway, S.L.; Jongens, T.A. Using Drosophila as a tool to identify Pharmacological Therapies for Fragile X Syndrome. Drug Discov. Today Technol. 2012, 10, e129–e136. [Google Scholar] [CrossRef] [Green Version]
- Schenck, A.; Bardoni, B.; Langmann, C.; Harden, N.; Mandel, J.L.; Giangrande, A. CYFIP/Sra-1 controls neuronal connectivity in Drosophila and links the Rac1 GTPase pathway to the fragile X protein. Neuron 2003, 38, 887–898. [Google Scholar] [CrossRef] [Green Version]
- Specchia, V.; Puricella, A.; D’Attis, S.; Massari, S.; Giangrande, A.; Bozzetti, M.P. Drosophila melanogaster as a Model to Study the Multiple Phenotypes, Related to Genome Stability of the Fragile-X Syndrome. Front. Genet. 2019, 10, 10. [Google Scholar] [CrossRef]
- Xu, K.; Bogert, B.A.; Li, W.; Su, K.; Lee, A.; Gao, F.B. The fragile X-related gene affects the crawling behavior of Drosophila larvae by regulating the mRNA level of the DEG/ENaC protein pickpocket1. Curr. Biol. 2004, 14, 1025–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Günther, M.N.; Nettesheim, G.; Shubeita, G.T. Quantifying and predicting Drosophila larvae crawling phenotypes. Sci. Rep. 2016, 6, 27972. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, A.; Price, J.L.; Man, B.; Young, M.W. Loss of circadian behavioral rhythms and per RNA oscillations in the Drosophila mutant timeless. Science 1994, 263, 1603–1606. [Google Scholar] [CrossRef]
- Bolduc, F.V.; Bell, K.; Cox, H.; Broadie, K.S.; Tully, T. Excess protein synthesis in Drosophila Fragile X mutants impairs long-term memory. Nat. Neurosci. 2008, 11, 1143–1145. [Google Scholar] [CrossRef]
- Bolduc, F.V.; Valente, D.; Nguyen, A.T.; Mitra, P.P.; Tully, T. An assay for social interaction in Drosophila fragile X mutants. Fly 2010, 4, 216–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gantois, I.; Popic, J.; Khoutorsky, A.; Sonenberg, N. Metformin for Treatment of Fragile X Syndrome and Other Neurological Disorders. Annu. Rev. Med. 2019, 70, 167–181. [Google Scholar] [CrossRef]
- Wirojanan, J.; Jacquemont, S.; Diaz, R.; Bacalman, S.; Anders, T.F.; Hagerman, R.J.; Goodlin-Jones, B.L. The efficacy of melatonin for sleep problems in children with autism, fragile X syndrome, or autism and fragile X syndrome. J. Clin. Sleep Med. 2009, 5, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Lumaban, J.G.; Nelson, D.L. The Fragile X proteins Fmrp and Fxr2p cooperate to regulate glucose metabolism in mice. Hum. Mol. Genet. 2015, 24, 2175–2184. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Poidevin, M.; Han, E.; Bi, J.; Jin, P. Circadian rhythm-dependent alterations of gene expression in Drosophila brain lacking fragile X mental retardation protein. PLoS ONE 2012, 7, e37937. [Google Scholar] [CrossRef] [Green Version]
- Bushey, D.; Tononi, G.; Cirelli, C. The Drosophila Fragile X Mental Retardation Gene Regulates Sleep Need. J. Neurosci. 2009, 29, 1948–1961. [Google Scholar] [CrossRef] [Green Version]
- Sekine, T.; Yamaguchi, T.; Hamano, K.; Siomi, H.; Saez, L.; Ishida, N.; Shimoda, M. Circadian phenotypes of Drosophila fragile x mutants in alternative genetic backgrounds. Zoolog. Sci. 2008, 25, 561–571. [Google Scholar] [CrossRef] [PubMed]
- McBride, S.M.; Bell, A.J.; Jongens, T.A. Behavior in a Drosophila Model of Fragile X. In Modeling Fragile X Syndrome; Denman, R.B., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 54, pp. 83–117. [Google Scholar]
- Andrew, D.R.; Moe, M.E.; Chen, D.; Tello, J.A.; Doser, R.L.; Conner, W.E.; Ghuman, J.K.; Restifo, L.L. Spontaneous motor-behavior abnormalities in two Drosophila models of neurodevelopmental disorders. J. Neurogenet. 2021, 35, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Van Alphen, B.; Yap, M.H.; Kirszenblat, L.; Kottler, B.; van Swinderen, B. A dynamic deep sleep stage in Drosophila. J. Neurosci. 2013, 33, 6917–6927. [Google Scholar] [CrossRef] [Green Version]
- Salcedo-Arellano, M.J.; Dufour, B.; McLennan, Y.; Martinez-Cerdeno, V.; Hagerman, R. Fragile X syndrome and associated disorders: Clinical aspects and pathology. Neurobiol. Dis. 2020, 136, 104740. [Google Scholar] [CrossRef] [PubMed]
- Kashima, R.; Redmond, P.L.; Ghatpande, P.; Roy, S.; Kornberg, T.B.; Hanke, T.; Knapp, S.; Lagna, G.; Hata, A. Hyperactive locomotion in a Drosophila model is a functional readout for the synaptic abnormalities underlying fragile X syndrome. Sci. Signal 2017, 10, eaai8133. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.R.; Kanellopoulos, A.K.; Bagni, C. Learning and behavioral deficits associated with the absence of the fragile X mental retardation protein: What a fly and mouse model can teach us. Learn. Mem. 2014, 21, 543–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, V.G.; Javadi, C.S.; Ngo, E.; Ngo, L.; Lagow, R.D.; Zhang, B. Age-related changes in climbing behavior and neural circuit physiology in Drosophila. Dev. Neurobiol. 2007, 67, 778–791. [Google Scholar] [CrossRef]
- Hagerman, P.J. The fragile X prevalence paradox. J. Med. Genet. 2008, 45, 498–499. [Google Scholar] [CrossRef]
- Volkmar, F.R.; Pauls, D. Autism. Lancet 2003, 362, 1133–1141. [Google Scholar] [CrossRef]
- Tauber, J.M.; Vanlandingham, P.A.; Zhang, B. Elevated Levels of the Vesicular Monoamine Transporter and a Novel Repetitive Behavior in the Drosophila Model of Fragile X Syndrome. PLoS ONE 2011, 6, e27100. [Google Scholar] [CrossRef] [Green Version]
- Lough, E.; Hanley, M.; Rodgers, J.; South, M.; Kirk, H.; Kennedy, D.P.; Riby, D.M. Violations of Personal Space in Young People with Autism Spectrum Disorders and Williams Syndrome: Insights from the Social Responsiveness Scale. J. Autism. Dev. Disord. 2015, 45, 4101–4108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, C.H.; McBride, S.M.; Schoenfeld, B.P.; Liebelt, D.A.; Ferreiro, D.; Ferrick, N.J.; Hinchey, P.; Kollaros, M.; Rudominer, R.L.; Terlizzi, A.M.; et al. Age-dependent cognitive impairment in a Drosophila fragile X model and its pharmacological rescue. Biogerontology 2010, 11, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Schoenfeld, B.P.; Bell, A.J.; Choi, C.H.; Bradley, M.P.; Hinchey, P.; Kollaros, M.; Park, J.H.; McBride, S.M.; Dockendorff, T.C. Short- and long-term memory are modulated by multiple isoforms of the fragile X mental retardation protein. J. Neurosci. 2010, 30, 6782–6792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, S.M.; Choi, C.H.; Wang, Y.; Liebelt, D.; Braunstein, E.; Ferreiro, D.; Sehgal, A.; Siwicki, K.K.; Dockendorff, T.C.; Nguyen, H.T.; et al. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron 2005, 45, 753–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.; Bray, S.M.; Li, Z.; Zarnescu, D.C.; He, C.; Jin, P.; Warren, S.T. Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat. Chem. Biol. 2008, 4, 256–263. [Google Scholar] [CrossRef]
- Hall, J.C. The mating of a fly. Science 1994, 264, 1702–1714. [Google Scholar] [CrossRef]
- Kim, K.; Hessl, D.; Randol, J.L.; Espinal, G.M.; Schneider, A.; Protic, D.; Aydin, E.Y.; Hagerman, R.J.; Hagerman, P.J. Association between IQ and FMR1 protein (FMRP) across the spectrum of CGG repeat expansions. PLoS ONE 2019, 14, e0226811. [Google Scholar] [CrossRef] [Green Version]
- Budimirovic, D.B.; Schlageter, A.; Filipovic-Sadic, S.; Protic, D.D.; Bram, E.; Mahone, E.M.; Nicholson, K.; Culp, K.; Javanmardi, K.; Kemppainen, J.; et al. A Genotype-Phenotype Study of High-Resolution FMR1 Nucleic Acid and Protein Analyses in Fragile X Patients with Neurobehavioral Assessments. Brain Sci. 2020, 10, 694. [Google Scholar] [CrossRef]
- Coffee, R.L., Jr.; Williamson, A.J.; Adkins, C.M.; Gray, M.C.; Page, T.L.; Broadie, K. In vivo neuronal function of the fragile X mental retardation protein is regulated by phosphorylation. Hum. Mol. Genet. 2012, 21, 900–915. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, S.D.; Welt, C.; Sherman, S. FMR1 and the continuum of primary ovarian insufficiency. Semin. Reprod. Med. 2011, 29, 299–307. [Google Scholar] [CrossRef]
- Hagerman, P. Fragile X-associated tremor/ataxia syndrome (FXTAS): Pathology and mechanisms. Acta Neuropathol. 2013, 126, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J.; Leehey, M.; Heinrichs, W.; Tassone, F.; Wilson, R.; Hills, J.; Grigsby, J.; Gage, B.; Hagerman, P.J. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 2001, 57, 127–130. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Protic, D.; Rajaratnam, A.; Salcedo-Arellano, M.J.; Aydin, E.Y.; Schneider, A. Fragile X-Associated Neuropsychiatric Disorders (FXAND). Front. Psychiatry 2018, 9, 564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabal-Herrera, A.M.; Tassanakijpanich, N.; Salcedo-Arellano, M.J.; Hagerman, R.J. Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS): Pathophysiology and Clinical Implications. Int. J. Mol. Sci. 2020, 21, 4391. [Google Scholar] [CrossRef] [PubMed]
- Jacquemont, S.; Hagerman, R.J.; Leehey, M.A.; Hall, D.A.; Levine, R.A.; Brunberg, J.A.; Zhang, L.; Jardini, T.; Gane, L.W.; Harris, S.W.; et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA 2004, 291, 460–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giulivi, C.; Napoli, E.; Tassone, F.; Halmai, J.; Hagerman, R. Plasma metabolic profile delineates roles for neurodegeneration, pro-inflammatory damage and mitochondrial dysfunction in the FMR1 premutation. Biochem. J. 2016, 473, 3871–3888. [Google Scholar] [CrossRef]
- Jin, P.; Zarnescu, D.C.; Zhang, F.; Pearson, C.E.; Lucchesi, J.C.; Moses, K.; Warren, S.T. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron 2003, 39, 739–747. [Google Scholar] [CrossRef] [Green Version]
- Qurashi, A.; Liu, H.; Ray, L.; Nelson, D.L.; Duan, R.; Jin, P. Chemical screen reveals small molecules suppressing fragile X premutation rCGG repeat-mediated neurodegeneration in Drosophila. Hum. Mol. Genet. 2012, 21, 2068–2075. [Google Scholar] [CrossRef] [Green Version]
- Todd, P.K.; Oh, S.Y.; Krans, A.; He, F.; Sellier, C.; Frazer, M.; Renoux, A.J.; Chen, K.C.; Scaglione, K.M.; Basrur, V.; et al. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 2013, 78, 440–455. [Google Scholar] [CrossRef] [Green Version]
- Jin, P.; Duan, R.; Qurashi, A.; Qin, Y.; Tian, D.; Rosser, T.C.; Liu, H.; Feng, Y.; Warren, S.T. Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron 2007, 55, 556–564. [Google Scholar] [CrossRef] [Green Version]
- Sofola, O.A.; Jin, P.; Qin, Y.; Duan, R.; Liu, H.; de Haro, M.; Nelson, D.L.; Botas, J. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron 2007, 55, 565–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leigh, M.J.; Nguyen, D.V.; Mu, Y.; Winarni, T.I.; Schneider, A.; Chechi, T.; Polussa, J.; Doucet, P.; Tassone, F.; Rivera, S.M.; et al. A randomized double-blind, placebo-controlled trial of minocycline in children and adolescents with fragile x syndrome. J. Dev. Behav. Pediatr. 2013, 34, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Monyak, R.E.; Emerson, D.; Schoenfeld, B.P.; Zheng, X.; Chambers, D.B.; Rosenfelt, C.; Langer, S.; Hinchey, P.; Choi, C.H.; McDonald, T.V.; et al. Insulin signaling misregulation underlies circadian and cognitive deficits in a Drosophila fragile X model. Mol. Psychiatry 2017, 22, 1140–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biag, H.M.B.; Potter, L.A.; Wilkins, V.; Afzal, S.; Rosvall, A.; Salcedo-Arellano, M.J.; Rajaratnam, A.; Manzano-Nunez, R.; Schneider, A.; Tassone, F.; et al. Metformin treatment in young children with fragile X syndrome. Mol. Genet. Genomic Med. 2019, 7, e956. [Google Scholar] [CrossRef] [Green Version]
- Dy, A.B.C.; Tassone, F.; Eldeeb, M.; Salcedo-Arellano, M.J.; Tartaglia, N.; Hagerman, R. Metformin as targeted treatment in fragile X syndrome. Clin. Genet. 2018, 93, 216–222. [Google Scholar] [CrossRef]
- Protic, D.; Aydin, E.Y.; Tassone, F.; Tan, M.M.; Hagerman, R.J.; Schneider, A. Cognitive and behavioral improvement in adults with fragile X syndrome treated with metformin-two cases. Mol. Genet. Genomic Med. 2019, 7, e00745. [Google Scholar] [CrossRef]
- Protic, D.; Kaluzhny, P.; Tassone, F.; Hagerman, R. Prepubertal Metformin Treatment in Fragile X Syndrome Alleviated Macroorchidism: A Case Study. Adv. Clin. Transl. Res. 2019, 3, 100021. [Google Scholar]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trajković, J.; Makevic, V.; Pesic, M.; Pavković-Lučić, S.; Milojevic, S.; Cvjetkovic, S.; Hagerman, R.; Budimirovic, D.B.; Protic, D. Drosophila melanogaster as a Model to Study Fragile X-Associated Disorders. Genes 2023, 14, 87. https://doi.org/10.3390/genes14010087
Trajković J, Makevic V, Pesic M, Pavković-Lučić S, Milojevic S, Cvjetkovic S, Hagerman R, Budimirovic DB, Protic D. Drosophila melanogaster as a Model to Study Fragile X-Associated Disorders. Genes. 2023; 14(1):87. https://doi.org/10.3390/genes14010087
Chicago/Turabian StyleTrajković, Jelena, Vedrana Makevic, Milica Pesic, Sofija Pavković-Lučić, Sara Milojevic, Smiljana Cvjetkovic, Randi Hagerman, Dejan B. Budimirovic, and Dragana Protic. 2023. "Drosophila melanogaster as a Model to Study Fragile X-Associated Disorders" Genes 14, no. 1: 87. https://doi.org/10.3390/genes14010087