
Methodological Changes in the Field of Paleogenetics
 , , , , and
, , , , and
Abstract
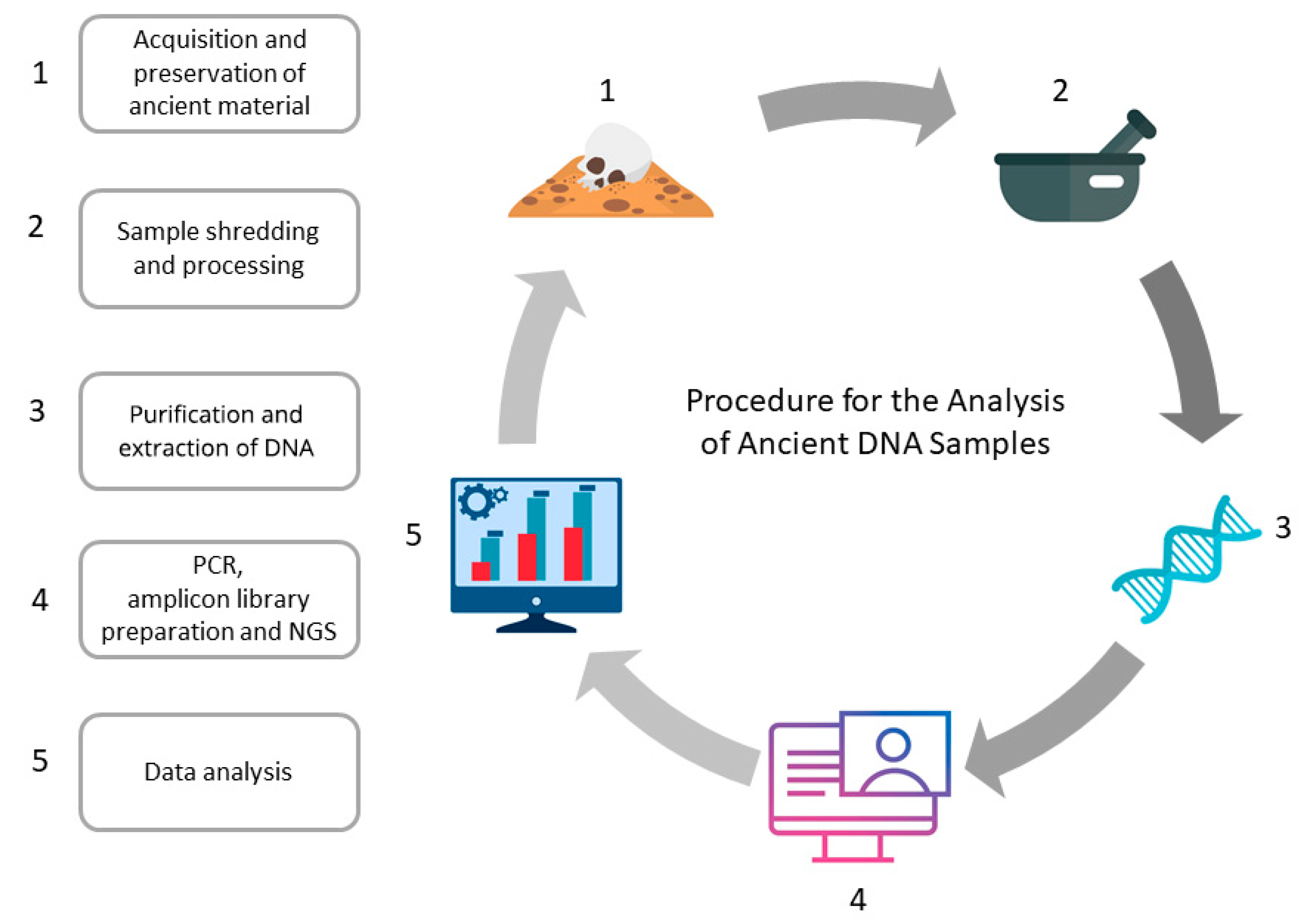
:1. Introduction
2. History of aDNA Research
3. Damage of aDNA
4. Materials, Methods and Contamination
5. Extraction of Ancient DNA
6. Amplification
7. Target Enrichment via Hybridization-Based Capture
8. Sequencing
9. Where Paleogenetic and Forensic Sciences Converge
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Paabo, S. Ancient DNA: Extraction, Characterization, Molecular Cloning, and Enzymatic Amplification. Proc. Natl. Acad. Sci. USA 1989, 86, 1939–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higuchi, R.; Bowman, B.; Freiberger, M.; Ryder, O.A.; Wilson, A.C. DNA Sequences from the Quagga, an Extinct Member of the Horse Family. Nature 1984, 312, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Ancient DNA|Definition of Ancient DNA by Medical Dictionary. Available online: https://medical-dictionary.thefreedictionary.com/Ancient+DNA (accessed on 6 January 2023).
- Barlow, A.; Cahill, J.A.; Hartmann, S.; Theunert, C.; Xenikoudakis, G.; Fortes, G.G.; Paijmans, J.L.A.; Rabeder, G.; Frischauf, C.; Grandal-d’Anglade, A.; et al. Partial Genomic Survival of Cave Bears in Living Brown Bears. Nat. Ecol. Evol. 2018, 2, 1563–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.D.E.; Magee, D.A.; McGettigan, P.A.; Teasdale, M.D.; Edwards, C.J.; Lohan, A.J.; Murphy, A.; Braud, M.; Donoghue, M.T.; Liu, Y.; et al. Genome Sequencing of the Extinct Eurasian Wild Aurochs, Bos Primigenius, Illuminates the Phylogeography and Evolution of Cattle. Genome Biol. 2015, 16, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palkopoulou, E.; Mallick, S.; Skoglund, P.; Enk, J.; Rohland, N.; Li, H.; Omrak, A.; Vartanyan, S.; Poinar, H.; Götherström, A.; et al. Complete Genomes Reveal Signatures of Demographic and Genetic Declines in the Woolly Mammoth. Curr. Biol. 2015, 25, 1395–1400. [Google Scholar] [CrossRef] [Green Version]
- Wielgus, K.; Danielewski, M.; Walkowiak, J. Svante Pääbo, Reader of the Neanderthal Genome. Acta Physiol. 2022, 237, e13902. [Google Scholar] [CrossRef]
- Mathieson, I.; Lazaridis, I.; Rohland, N.; Mallick, S.; Patterson, N.; Roodenberg, S.A.; Harney, E.; Stewardson, K.; Fernandes, D.; Novak, M.; et al. Genome-Wide Patterns of Selection in 230 Ancient Eurasians. Nature 2015, 528, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Klunk, J.; Vilgalys, T.P.; Demeure, C.E.; Cheng, X.; Shiratori, M.; Madej, J.; Beau, R.; Elli, D.; Patino, M.I.; Redfern, R.; et al. Evolution of Immune Genes Is Associated with the Black Death. Nature 2022, 611, 312–319. [Google Scholar] [CrossRef]
- Benton, M.L.; Abraham, A.; LaBella, A.L.; Abbot, P.; Rokas, A.; Capra, J.A. The Influence of Evolutionary History on Human Health and Disease. Nat. Rev. Genet. 2021, 22, 269–283. [Google Scholar] [CrossRef]
- Quintana-Murci, L. Understanding Rare and Common Diseases in the Context of Human Evolution. Genome Biol. 2016, 17, 225. [Google Scholar] [CrossRef]
- Harney, É.; May, H.; Shalem, D.; Rohland, N.; Mallick, S.; Lazaridis, I.; Sarig, R.; Stewardson, K.; Nordenfelt, S.; Patterson, N.; et al. Ancient DNA from Chalcolithic Israel Reveals the Role of Population Mixture in Cultural Transformation. Nat. Commun. 2018, 9, 3336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, G.; Haak, W.; Adler, C.J.; Roth, C.; Szécsényi-Nagy, A.; Karimnia, S.; Möller-Rieker, S.; Meller, H.; Ganslmeier, R.; Friederich, S.; et al. Ancient DNA Reveals Key Stages in the Formation of Central European Mitochondrial Genetic Diversity. Science 2013, 342, 257–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murchie, T.J.; Monteath, A.J.; Mahony, M.E.; Long, G.S.; Cocker, S.; Sadoway, T.; Karpinski, E.; Zazula, G.; MacPhee, R.D.E.; Froese, D.; et al. Collapse of the Mammoth-Steppe in Central Yukon as Revealed by Ancient Environmental DNA. Nat. Commun. 2021, 12, 7120. [Google Scholar] [CrossRef] [PubMed]
- Ottoni, C.; Bekaert, B.; Decorte, R. DNA Degradation: Current Knowledge and Progress in DNA Analysis. In Taphonomy of Human Remains: Forensic Analysis of the Dead and the Depositional Environment; John Wiley & Sons, Ltd.: New York, NY, USA, 2017; pp. 65–80. [Google Scholar]
- Pääbo, S.; Wilson, A.C. Polymerase Chain Reaction Reveals Cloning Artefacts. Nature 1988, 334, 387–388. [Google Scholar] [CrossRef]
- Gitschier, J. Imagine: An Interview with Svante Pääbo. PLoS Genet. 2008, 4, e1000035. [Google Scholar] [CrossRef]
- Willerslev, E.; Cooper, A. Ancient DNA. Proc. R. Soc. B Biol. Sci. 2005, 272, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Jones, E.D.; Bösl, E. Ancient Human DNA: A History of Hype (Then and Now). J. Soc. Archaeol. 2021, 21, 236–255. [Google Scholar] [CrossRef]
- Poinar, H.N.; Schwarz, C.; Qi, J.; Shapiro, B.; MacPhee, R.D.E.; Buigues, B.; Tikhonov, A.; Huson, D.M.; Tomsho, L.P.; Auch, A.; et al. Metagenomics to Paleogenomics: Large-Scale Sequencing of Mammoth DNA. Science 2006, 311, 392–394. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, M.; Li, Y.; Lindgreen, S.; Pedersen, J.S.; Albrechtsen, A.; Moltke, I.; Metspalu, M.; Metspalu, E.; Kivisild, T.; Gupta, R.; et al. Ancient Human Genome Sequence of an Extinct Palaeo-Eskimo. Nature 2010, 463, 757–762. [Google Scholar] [CrossRef] [Green Version]
- Green, R.E.; Krause, J.; Briggs, A.W.; Maricic, T.; Stenzel, U.; Kircher, M.; Patterson, N.; Li, H.; Zhai, W.; Fritz, M.H.Y.; et al. A Draft Sequence of the Neandertal Genome. Science 2010, 328, 710–722. [Google Scholar] [CrossRef]
- Reich, D.; Green, R.E.; Kircher, M.; Krause, J.; Patterson, N.; Durand, E.Y.; Viola, B.; Briggs, A.W.; Stenzel, U.; Johnson, P.L.F.; et al. Genetic History of an Archaic Hominin Group from Denisova Cave in Siberia. Nature 2010, 468, 1053–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjær, K.H.; Pedersen, M.W.; de Sanctis, B.; de Cahsan, B.; Korneliussen, T.S.; Michelsen, C.S.; Sand, K.K.; Jelavić, S.; Ruter, A.H.; Schmidt, A.M.A.; et al. A 2-Million-Year-Old Ecosystem in Greenland Uncovered by Environmental DNA. Nature 2022, 612, 283–291. [Google Scholar] [CrossRef]
- van der Valk, T.; Pečnerová, P.; Díez-del-Molino, D.; Bergström, A.; Oppenheimer, J.; Hartmann, S.; Xenikoudakis, G.; Thomas, J.A.; Dehasque, M.; Sağlıcan, E.; et al. Million-Year-Old DNA Sheds Light on the Genomic History of Mammoths. Nature 2021, 591, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, D.A.; Dickel, C.D.; Hauswirth, W.W.; Parham, P. Ancient HLA Genes from 7500-Year-Old Archaeological Remains. Nature 1991, 349, 785–788. [Google Scholar] [CrossRef]
- Poinar, H.N.; Hofreiter, M.; Spaulding, W.G.; Martin, P.S.; Stankiewicz, B.A.; Bland, H.; Evershed, R.P.; Possnert, G.; Pääbo, S. Molecular Coproscopy: Dung and Diet of the Extinct Ground Sloth Nothrotheriops Shastensis. Science 1998, 281, 402–406. [Google Scholar] [CrossRef] [Green Version]
- Willerslev, E.; Hansen, A.J.; Binladen, J.; Brand, T.B.; Gilbert, M.T.P.; Shapiro, B.; Bunce, M.; Wiuf, C.; Gilichinsky, D.A.; Cooper, A. Diverse Plant and Animal Genetic Records from Holocene and Pleistocene Sediments. Science 2003, 300, 791–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haak, W.; Forster, P.; Bramanti, B.; Matsumura, S.; Brandt, G.; Tänzer, M.; Villems, R.; Renfrew, C.; Gronenborn, D.; Alt, K.W.; et al. Ancient DNA from the First European Farmers in 7500-Year-Old Neolithic Sites. Science 2005, 310, 1016–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, W.; Drautz, D.I.; Ratan, A.; Pusey, B.; Qi, J.; Lesk, A.M.; Tomsho, L.P.; Packard, M.D.; Zhao, F.; Sher, A.; et al. Sequencing the Nuclear Genome of the Extinct Woolly Mammoth. Nature 2008, 456, 387–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, R.E.; Malaspinas, A.S.; Krause, J.; Briggs, A.W.; Johnson, P.L.F.; Uhler, C.; Meyer, M.; Good, J.M.; Maricic, T.; Stenzel, U.; et al. A Complete Neandertal Mitochondrial Genome Sequence Determined by High-Throughput Sequencing. Cell 2008, 134, 416–426. [Google Scholar] [CrossRef] [Green Version]
- Bos, K.I.; Schuenemann, V.J.; Golding, G.B.; Burbano, H.A.; Waglechner, N.; Coombes, B.K.; McPhee, J.B.; Dewitte, S.N.; Meyer, M.; Schmedes, S.; et al. A Draft Genome of Yersinia Pestis from Victims of the Black Death. Nature 2011, 478, 506–510. [Google Scholar] [CrossRef]
- Meyer, M.; Kircher, M.; Gansauge, M.T.; Li, H.; Racimo, F.; Mallick, S.; Schraiber, J.G.; Jay, F.; Prüfer, K.; de Filippo, C.; et al. A High-Coverage Genome Sequence from an Archaic Denisovan Individual. Science 2012, 338, 222–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, A.; Graefen, A.; Ball, M.; Matzas, M.; Boisguerin, V.; Maixner, F.; Leidinger, P.; Backes, C.; Khairat, R.; Forster, M.; et al. New Insights into the Tyrolean Iceman’s Origin and Phenotype as Inferred by Whole-Genome Sequencing. Nat. Commun. 2012, 3, 698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warinner, C.; Rodrigues, J.F.M.; Vyas, R.; Trachsel, C.; Shved, N.; Grossmann, J.; Radini, A.; Hancock, Y.; Tito, R.Y.; Fiddyment, S.; et al. Pathogens and Host Immunity in the Ancient Human Oral Cavity. Nat. Genet. 2014, 46, 336–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, T.E.; Fortes, G.G.; Balaresque, P.; Thomas, M.G.; Balding, D.; Delser, P.M.; Neumann, R.; Parson, W.; Knapp, M.; Walsh, S.; et al. Identification of the Remains of King Richard III. Nat. Commun. 2014, 5, 5631. [Google Scholar] [CrossRef] [Green Version]
- Prüfer, K.; Racimo, F.; Patterson, N.; Jay, F.; Sankararaman, S.; Sawyer, S.; Heinze, A.; Renaud, G.; Sudmant, P.H.; de Filippo, C.; et al. The Complete Genome Sequence of a Neanderthal from the Altai Mountains. Nature 2014, 505, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prüfer, K.; de Filippo, C.; Grote, S.; Mafessoni, F.; Korlević, P.; Hajdinjak, M.; Vernot, B.; Skov, L.; Hsieh, P.; Peyrégne, S.; et al. A High-Coverage Neandertal Genome from Vindija Cave in Croatia. Science 2017, 358, 655–658. [Google Scholar] [CrossRef] [Green Version]
- Margaryan, A.; Lawson, D.J.; Sikora, M.; Racimo, F.; Rasmussen, S.; Moltke, I.; Cassidy, L.M.; Jørsboe, E.; Ingason, A.; Pedersen, M.W.; et al. Population Genomics of the Viking World. Nature 2020, 585, 390–396. [Google Scholar] [CrossRef]
- Mafessoni, F.; Grote, S.; de Filippo, C.; Slon, V.; Kolobova, K.A.; Viola, B.; Markin, S.V.; Chintalapati, M.; Peyrégne, S.; Skov, L.; et al. A High-Coverage Neandertal Genome from Chagyrskaya Cave. Proc. Natl. Acad. Sci. USA 2020, 117, 15132–15136. [Google Scholar] [CrossRef]
- Bailly, V.; Verly, W.G. Possible Roles of F-Elimination and 6-Elimination Reactions in the Repair of DNA Containing AP (Apurinic/Apyrimidinic) Sites in Mammalian Cells. Biochem. J. 1988, 253, 553–559. [Google Scholar] [CrossRef]
- Overballe-Petersen, S.; Orlando, L.; Willerslev, E. Next-Generation Sequencing Offers New Insights into DNA Degradation. Trends Biotechnol. 2012, 30, 364–368. [Google Scholar] [CrossRef]
- Lindahl, T.; Karlström, O. Heat-Induced Depyrimidination of Deoxyribonucleic Acid in Neutral Solution. Biochemistry 1973, 12, 5151–5154. [Google Scholar] [CrossRef]
- Lindahl, T.; Nyberg, B. Rate of Depurination of Native Deoxyribonucleic Acid. Biochemistry 1972, 11, 3610–3618. [Google Scholar] [CrossRef]
- Shapiro, R. Damage to DNA Caused by Hydrolysis. In Chromosome Damage and Repair; Springer: New York, NY, USA, 1981; pp. 3–18. [Google Scholar]
- Poulakakis, N.; Tselikas, A.; Bitsakis, I.; Mylonas, M.; Lymberakis, P. Ancient DNA and the Genetic Signature of Ancient Greek Manuscripts. J. Archaeol. Sci. 2007, 34, 675–680. [Google Scholar] [CrossRef]
- Briggs, A.W.; Stenzel, U.; Johnson, P.L.F.; Green, R.E.; Kelso, J.; Prüfer, K.; Meyer, M.; Krause, J.; Ronan, M.T.; Lachmann, M.; et al. Patterns of Damage in Genomic DNA Sequences from a Neandertal. Proc. Natl. Acad. Sci. USA 2007, 104, 14616–14621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabney, J.; Meyer, M.; Pääbo, S. Ancient DNA Damage. Cold Spring Harb. Perspect. Biol. 2013, 5, a012567. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and Decay of the Primary Structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Pääbo, S.; Gifford, J.A.; Wilson, A.C. Mitochondrial DNA Sequences from a 7000-Year Old Brain. Nucleic Acids Res. 1988, 16, 9775–9787. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.T.P.; Menez, L.; Janaway, R.C.; Tobin, D.J.; Cooper, A.; Wilson, A.S. Resistance of Degraded Hair Shafts to Contaminant DNA. Forensic Sci. Int. 2006, 156, 208–212. [Google Scholar] [CrossRef]
- Gilbert, M.T.P.; Wilson, A.S.; Bunce, M.; Hansen, A.J.; Willerslev, E.; Shapiro, B.; Higham, T.F.G.; Richards, M.P.; O’Connell, T.C.; Tobin, D.J.; et al. Ancient Mitochondrial DNA from Hair. Curr. Biol. 2004, 14, R463–R464. [Google Scholar] [CrossRef]
- Wolinsky, H. History in a Single Hair. EMBO Rep. 2010, 11, 427–430. [Google Scholar] [CrossRef]
- Bonnichsen, R.; Hodges, L.; Ream, W.; Field, K.G.; Kirner, D.L.; Selsor, K.; Taylor, R.E. Methods for the Study of Ancient Hair: Radiocarbon Dates and Gene Sequences from Individual Hairs. J. Archaeol. Sci. 2001, 28, 775–785. [Google Scholar] [CrossRef]
- Wadsworth, C.; Procopio, N.; Anderung, C.; Carretero, J.M.; Iriarte, E.; Valdiosera, C.; Elburg, R.; Penkman, K.; Buckley, M. Comparing Ancient DNA Survival and Proteome Content in 69 Archaeological Cattle Tooth and Bone Samples from Multiple European Sites. J. Proteom. 2017, 158, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Latham, K.E.; Miller, J.J. DNA Recovery and Analysis from Skeletal Material in Modern Forensic Contexts. Forensic Sci. Res. 2019, 4, 51–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamba, C.; Jones, E.R.; Teasdale, M.D.; McLaughlin, R.L.; Gonzalez-Fortes, G.; Mattiangeli, V.; Domboróczki, L.; Kovári, I.; Pap, I.; Anders, A.; et al. Genome Flux and Stasis in a Five Millennium Transect of European Prehistory. Nat. Commun. 2014, 5, 5257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, A.E.; Sabin, S.; Ziesemer, K.; Vågene, Å.J.; Schroeder, H.; Ozga, A.T.; Sankaranarayanan, K.; Hofman, C.A.; Fellows Yates, J.A.; Salazar-García, D.C.; et al. Differential Preservation of Endogenous Human and Microbial DNA in Dental Calculus and Dentin. Sci. Rep. 2018, 8, 9822. [Google Scholar] [CrossRef]
- Zhang, D.; Xia, H.; Chen, F.; Li, B.; Slon, V.; Cheng, T.; Yang, R.; Jacobs, Z.; Dai, Q.; Massilani, D.; et al. Denisovan DNA in Late Pleistocene Sediments from Baishiya Karst Cave on the Tibetan Plateau. Science 2020, 370, 584–587. [Google Scholar] [CrossRef]
- Llamas, B.; Valverde, G.; Fehren-Schmitz, L.; Weyrich, L.S.; Cooper, A.; Haak, W. From the Field to the Laboratory: Controlling DNA Contamination in Human Ancient DNA Research in the High-Throughput Sequencing Era. Sci. Technol. Archaeol. Res. 2017, 3, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.; Poinar, H.N. Ancient DNA: Do It Right or Not at All. Science 2000, 289, 1139. [Google Scholar] [CrossRef]
- Orlando, L.; Allaby, R.; Skoglund, P.; der Sarkissian, C.; Stockhammer, P.W.; Ávila-Arcos, M.C.; Fu, Q.; Krause, J.; Willerslev, E.; Stone, A.C.; et al. Ancient DNA Analysis. Nat. Rev. Methods Prim. 2021, 1, 14. [Google Scholar] [CrossRef]
- Poinar, H.N. The Top 10 List: Criteria of Authenticity for DNA from Ancient and Forensic Samples. Int. Congr. Ser. 2003, 1239, 575–579. [Google Scholar] [CrossRef]
- Marchi, N.; Winkelbach, L.; Schulz, I.; Brami, M.; Hofmanová, Z.; Blöcher, J.; Reyna-Blanco, C.S.; Diekmann, Y.; Thiéry, A.; Kapopoulou, A.; et al. The Genomic Origins of the World’s First Farmers. Cell 2022, 185, 1842−1859.e18. [Google Scholar] [CrossRef] [PubMed]
- Rohland, N.; Hofreiter, M. Ancient DNA Extraction from Bones and Teeth. Nat. Protoc. 2007, 2, 1756–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xavier, C.; Eduardoff, M.; Bertoglio, B.; Amory, C.; Berger, C.; Casas-Vargas, A.; Pallua, J.; Parson, W. Evaluation of DNA Extraction Methods Developed for Forensic and Ancient DNA Applications Using Bone Samples of Different Age. Genes 2021, 12, 146. [Google Scholar] [CrossRef] [PubMed]
- Nieves-Colón, M.A.; Ozga, A.T.; Pestle, W.J.; Cucina, A.; Tiesler, V.; Stanton, T.W.; Stone, A.C. Comparison of Two Ancient DNA Extraction Protocols for Skeletal Remains from Tropical Environments. Am. J. Phys. Anthropol. 2018, 166, 824–836. [Google Scholar] [CrossRef] [PubMed]
- Höss, M.; Pääbo, S. DNA Extraction from Pleistocene Bones by a Silica-Based Purification Method. Nucleic Acids Res. 1993, 21, 3913–3914. [Google Scholar] [CrossRef] [Green Version]
- Zeyland, J.; Wolko; Bocianowski, J.; Szalata, M.; Słomski, R.; Dzieduszycki, A.M.; Ryba, M.; Przystałowska, H.; Lipiński, D. Complete Mitochondrial Genome of Wild Aurochs (Bos Primigenius) Reconstructed from Ancient DNA. Pol. J. Vet. Sci. 2013, 16, 265–273. [Google Scholar] [CrossRef] [Green Version]
- Juras, A. 2012. Etnogeneza Słowian w Świetle Badań Kopalnego DNA. Ph.D. Thesis, Adam Mickiewicz University, Poznan, Poland.
- Loreille, O.M.; Diegoli, T.M.; Irwin, J.A.; Coble, M.D.; Parsons, T.J. High Efficiency DNA Extraction from Bone by Total Demineralization. Forensic Sci. Int. Genet. 2007, 1, 191–195. [Google Scholar] [CrossRef]
- Dabney, J.; Meyer, M. Extraction of Highly Degraded DNA from Ancient Bones and Teeth. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2019; Volume 1963, pp. 25–29. [Google Scholar]
- Gamba, C.; Hanghøj, K.; Gaunitz, C.; Alfarhan, A.H.; Alquraishi, S.A.; Al-Rasheid, K.A.S.; Bradley, D.G.; Orlando, L. Comparing the Performance of Three Ancient DNA Extraction Methods for High-Throughput Sequencing. Mol. Ecol. Resour. 2016, 16, 459–469. [Google Scholar] [CrossRef]
- Boessenkool, S.; Hanghøj, K.; Nistelberger, H.M.; der Sarkissian, C.; Gondek, A.T.; Orlando, L.; Barrett, J.H.; Star, B. Combining Bleach and Mild Predigestion Improves Ancient DNA Recovery from Bones. Mol. Ecol. Resour. 2017, 17, 742–751. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.Y.; Eng, B.; Waye, J.S.; Dudar, J.C.; Saunders, S.R. Improved DNA Extraction from Ancient Bones Using Silica-Based Spin Columns. Am. J. Phys. Anthropol. 1998, 105, 539–543. [Google Scholar] [CrossRef]
- Wales, N.; Kistler, L. Extraction of Ancient DNA from Plant Remains. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2019; Volume 1963, pp. 45–55. [Google Scholar]
- Tuross, N. The Biochemistry of Ancient DNA in Bone. Experientia 1994, 50, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Brzobohatá, K.; Drozdová, E.; Smutný, J.; Zeman, T.; Beňuš, R. Comparison of Suitability of the Most Common Ancient DNA Quantification Methods. Genet. Test. Mol. Biomark. 2017, 21, 265–271. [Google Scholar] [CrossRef]
- Veeramah, K.R.; Hammer, M.F. The Impact of Whole-Genome Sequencing on the Reconstruction of Human Population History. Nat. Rev. Genet. 2014, 15, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Lipiński, D.; Przystałowska, H.; Szalata, M.; Zeyland, J.; Wielgus, K.; Frąckowiak, H.; Dzieduszycki, A.M.; Ryba, M.S.; Słomski, R. Biotechnology in the Restoration of Extinct Animal Species. An Analysis of Genomic and Mitochondrial DNA of Aurochs, BioTechnologia 2011, 92, 13–21. [Google Scholar]
- Lee, P.L.M.; Prys-Jones, R.P. Extracting DNA from Museum Bird Eggs, and Whole Genome Amplification of Archive DNA. Mol. Ecol. Resour. 2008, 8, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Delamoye, M.; Duverneuil, C.; Riva, K.; Leterreux, M.; Taieb, S.; de Mazancourt, P. False Homozygosities at Various Loci Revealed by Discrepancies between Commercial Kits: Implications for Genetic Databases. Forensic Sci. Int. 2004, 143, 47–52. [Google Scholar] [CrossRef]
- Harder, M.; Renneberg, R.; Meyer, P.; Krause-Kyora, B.; von Wurmb-Schwark, N. STR-Typing of Ancient Skeletal Remains: Which Multiplex-PCR Kit Is the Best. Croat. Med. J. 2012, 53, 416–422. [Google Scholar] [CrossRef] [Green Version]
- Tucker, V.C.; Kirkham, A.J.; Hopwood, A.J. Forensic Validation of the PowerPlex® ESI 16 STR Multiplex and Comparison of Performance with AmpFlSTR® SGM Plus. Int. J. Leg. Med. 2012, 126, 345–356. [Google Scholar] [CrossRef]
- Bini, C.; Cilli, E.; Sarno, S.; Traversari, M.; Fontani, F.; Boattini, A.; Pelotti, S.; Luiselli, D. Twenty-Seven Y-Chromosome Short Tandem Repeats Analysis of Italian Mummies of the 16th and 18th Centuries: An Interdisciplinary Research. Front. Genet. 2021, 12, 1629. [Google Scholar] [CrossRef]
- Gaudin, M.; Desnues, C. Hybrid Capture-Based Next Generation Sequencing and Its Application to Human Infectious Diseases. Front. Microbiol. 2018, 9, 2924. [Google Scholar] [CrossRef] [Green Version]
- Soares, A.E.R. Hybridization Capture of Ancient DNA Using RNA Baits. Methods Mol. Biol. 2019, 1963, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Scholl, U.I.; Ji, W.; Liu, T.; Tikhonova, I.R.; Zumbo, P.; Nayir, A.; Bakkaloǧlu, A.; Özen, S.; Sanjad, S.; et al. Genetic Diagnosis by Whole Exome Capture and Massively Parallel DNA Sequencing. Proc. Natl. Acad. Sci. USA 2009, 106, 19096–19101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, M.L.; Buenrostro, J.D.; Valdiosera, C.; Schroeder, H.; Allentoft, M.E.; Sikora, M.; Rasmussen, M.; Gravel, S.; Guillén, S.; Nekhrizov, G.; et al. Pulling out the 1%: Whole-Genome Capture for the Targeted Enrichment of Ancient DNA Sequencing Libraries. Am. J. Hum. Genet. 2013, 93, 852–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enk, J.M.; Devault, A.M.; Kuch, M.; Murgha, Y.E.; Rouillard, J.M.; Poinar, H.N. Ancient Whole Genome Enrichment Using Baits Built from Modern DNA. Mol. Biol. Evol. 2014, 31, 1292–1294. [Google Scholar] [CrossRef]
- Eduardoff, M.; Xavier, C.; Strobl, C.; Casas-Vargas, A.; Parson, W. Optimized MtDNA Control Region Primer Extension Capture Analysis for Forensically Relevant Samples and Highly Compromised MtDNA of Different Age and Origin. Genes 2017, 8, 237. [Google Scholar] [CrossRef] [Green Version]
- Loreille, O.; Ratnayake, S.; Bazinet, A.L.; Stockwell, T.B.; Sommer, D.D.; Rohland, N.; Mallick, S.; Johnson, P.L.F.; Skoglund, P.; Onorato, A.J.; et al. Biological Sexing of a 4000-Year-Old Egyptian Mummy Head to Assess the Potential of Nuclear DNA Recovery from the Most Damaged and Limited Forensic Specimens. Genes 2018, 9, 135. [Google Scholar] [CrossRef] [Green Version]
- Armbrecht, L.; Hallegraeff, G.; Bolch, C.J.S.; Woodward, C.; Cooper, A. Hybridisation Capture Allows DNA Damage Analysis of Ancient Marine Eukaryotes. Sci. Rep. 2021, 11, 3220. [Google Scholar] [CrossRef]
- Mohandesan, E.; Speller, C.F.; Peters, J.; Uerpmann, H.P.; Uerpmann, M.; de Cupere, B.; Hofreiter, M.; Burger, P.A. Combined Hybridization Capture and Shotgun Sequencing for Ancient DNA Analysis of Extinct Wild and Domestic Dromedary Camel. Mol. Ecol. Resour. 2017, 17, 300–313. [Google Scholar] [CrossRef] [Green Version]
- Schuenemann, V.J.; Bos, K.; DeWitte, S.; Schmedes, S.; Jamieson, J.; Mittnik, A.; Forrest, S.; Coombes, B.K.; Wood, J.W.; Earn, D.J.D.; et al. Targeted Enrichment of Ancient Pathogens Yielding the PPCP1 Plasmid of Yersinia Pestis from Victims of the Black Death. Proc. Natl. Acad. Sci. USA 2011, 108, E746–E752. [Google Scholar] [CrossRef] [Green Version]
- Feldman, M.; Harbeck, M.; Keller, M.; Spyrou, M.A.; Rott, A.; Trautmann, B.; Scholz, H.C.; Päffgen, B.; Peters, J.; McCormick, M.; et al. A High-Coverage Yersinia Pestis Genome from a Sixth-Century Justinianic Plague Victim. Mol. Biol. Evol. 2016, 33, 2911–2923. [Google Scholar] [CrossRef] [Green Version]
- Spyrou, M.A.; Tukhbatova, R.I.; Wang, C.C.; Valtueña, A.A.; Lankapalli, A.K.; Kondrashin, V.V.; Tsybin, V.A.; Khokhlov, A.; Kühnert, D.; Herbig, A.; et al. Analysis of 3800-Year-Old Yersinia Pestis Genomes Suggests Bronze Age Origin for Bubonic Plague. Nat. Commun. 2018, 9, 2234. [Google Scholar] [CrossRef] [PubMed]
- Mamanova, L.; Coffey, A.J.; Scott, C.E.; Kozarewa, I.; Turner, E.H.; Kumar, A.; Howard, E.; Shendure, J.; Turner, D.J. Target-Enrichment Strategies for next-Generation Sequencing. Nat. Methods 2010, 7, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-Generation Sequencing Technologies: An Overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef]
- Mardis, E.R. Next-Generation Sequencing Platforms. Annu. Rev. Anal. Chem. 2013, 6, 287–303. [Google Scholar] [CrossRef] [Green Version]
- Carøe, C.; Gopalakrishnan, S.; Vinner, L.; Mak, S.S.T.; Sinding, M.H.S.; Samaniego, J.A.; Wales, N.; Sicheritz-Pontén, T.; Gilbert, M.T.P. Single-Tube Library Preparation for Degraded DNA. Methods Ecol. Evol. 2018, 9, 410–419. [Google Scholar] [CrossRef] [Green Version]
- Gansauge, M.T.; Meyer, M. Single-Stranded DNA Library Preparation for the Sequencing of Ancient or Damaged DNA. Nat. Protoc. 2013, 8, 737–748. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, X.; Tang, P.S.; O’Leary, G.M.; Zhang, M. Targeted Sequencing of Both DNA Strands Barcoded and Captured Individually by RNA Probes to Identify Genome-Wide Ultra-Rare Mutations. Sci. Rep. 2017, 7, 3356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate Whole Human Genome Sequencing Using Reversible Terminator Chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Gansauge, M.T.; Gerber, T.; Glocke, I.; Korlević, P.; Lippik, L.; Nagel, S.; Riehl, L.M.; Schmidt, A.; Meyer, M. Single-Stranded DNA Library Preparation from Highly Degraded DNA Using T4 DNA Ligase. Nucleic Acids Res. 2017, 45, e79. [Google Scholar] [CrossRef] [Green Version]
- Barlow, A.; Fortes, G.M.G.; Dalen, L.; Pinhasi, R.; Gasparyan, B.; Rabeder, G.; Frischchauf, C.; Paijmans, J.L.A.; Hofreiter, M. Massive Influence of DNA Isolation and Library Preparation Approaches on Palaeogenomic Sequencing Data. BioRxiv 2016, 075911. [Google Scholar] [CrossRef] [Green Version]
- Sandoval-Velasco, M.; Lundstrøm, I.K.C.; Wales, N.; Ávila-Arcos, M.C.; Schroeder, H.; Gilbert, M.T.P. Relative Performance of Two DNA Extraction and Library Preparation Methods on Archaeological Human Teeth Samples. Sci. Technol. Archaeol. Res. 2017, 3, 80–88. [Google Scholar] [CrossRef]
- Zavala, E.I.; Thomas, J.T.; Sturk-Andreaggi, K.; Daniels-Higginbotham, J.; Meyers, K.K.; Barrit-Ross, S.; Aximu-Petri, A.; Richter, J.; Nickel, B.; Berg, G.E.; et al. Ancient DNA Methods Improve Forensic DNA Profiling of Korean War and World War II Unknowns. Genes 2022, 13, 129. [Google Scholar] [CrossRef] [PubMed]
- Pluta, D.; Lebioda, A.; Jonkisz, A.; Dobosz, T. First Successful DNA Isolation and Profiling from Bone Using an Approach That Is Non-Destructive toward Bone Surface. Arch. Med. Sądowej i Kryminol. 2016, 66, 65–70. [Google Scholar] [CrossRef]


{kind=link}
| Year of Discovery | Methods | Sample Material | Result | References |
|---|---|---|---|---|
| 1984 | Molecular cloning, Sanger’s sequencing | Dried muscle tissue, quagga specimen | Two sequenced mitochondrial DNA fragments (117 and 112 bp). First recovered aDNA. | [2] |
| 1988 | PCR | Dried muscle, quagga specimen | Detected cloning artefacts previously unnoticed in [2] with PCR. | [16] |
| 1988 | Molecular cloning, PCR | Numerous different ancient samples | Comparing the usefulness of molecular cloning and PCR in aDNA research. | [1] |
| 1991 | PCR | Human brain tissue, 6990–8130 years old | Sequenced fragments of 6 nuclear genes. | [26] |
| 1998 | PCR | Coprolite | Amplification of DNA from ancient feces. Analysis of the diet of the specimen and identification of species of the specimen. | [27] |
| 2003 | PCR | Sediment | First analysis of environmental aDNA | [28] |
| 2005 | PCR | Bones, teeth | Intact stretches of mitochondrial DNA from 24 Neolithic skeletons. | [29] |
| 2006 | NGS | Woolly mammoth’s mandible | 28 million bp sequenced, 13 million bp were endogenous. First use of NGS in paleogenetics. Analyses of the metagenomic nature of ancient remains. | [20] |
| 2008 | NGS | Woolly mammoth’s hair | 4.17 billion bp sequenced, 3.3 billion of which were endogenous | [30] |
| 2008 | NGS | Neanderthal bone | Fully sequenced Neanderthal mitochondrial genome | [31] |
| 2010 | NGS | 21 Neanderthal bones, 3 selected for further analysis | First sequenced Neanderthal genome (1.2× coverage), evidence for Neanderthals interbreeding with anatomically modern humans | [22] |
| 2010 | NGS | Finger bone | Discovery of Denisovans and sequenced Denisovan genome | [23] |
| 2010 | NGS | Hair | First sequenced ancient human genome (Paleo-Inuit) | [21] |
| 2011 | NGS | Teeth, bones | First fully sequenced genome of ancient bacterial pathogen | [32] |
| 2012 | NGS | Finger bone | First high coverage (30×) of Denisovan genome, use of single-stranded library preparation. | [33] |
| 2012 | NGS | Bone from the mummy of Tyrolean Iceman | Genome of Tyrolean Iceman fully sequenced, analysis of phenotype and metagenome | [34] |
| 2014 | NGS | Ancient calcified dental plaque | First high-resolution taxonomic and proteomic analysis of ancient oral microbiome from calcified dental plaque | [35] |
| 2014 | NGS | Bones | Identification of English king Richard III | [36] |
| 2014 | NGS | Toe phalanx | High-quality sequence of Neanderthal woman genome (coverage ~50×) | [37] |
| 2015 | NGS | - | Analysis of 230 ancient Eurasian genomes to determine genome-wide patterns of selection | [8] |
| 2015 | NGS | Molar tooth, soft tissue | Complete high-quality two woolly mammoth genomes, analysis of demographic history | [6] |
| 2015 | NGS | Auroch bone | 6750-year-old auroch genome, analysis of domestication process and its impact on the genome | [5] |
| 2017 | NGS | Bone | High-coverage genome (30×) of Neanderthal from Vindija Cave, analysis of gene flow between Neanderthals, Denisovans and anatomically modern humans | [38] |
| 2020 | NGS | - | Sequencing of 442 genomes from archaeological sites across Europe and Greenland to understand the expansion of the Scandinavian population during the Viking Age | [39] |
| 2020 | NGS | Finger bone | High coverage (27×) sequencing of a Neanderthal from Chagyrskaya Cave. Detection of selection patterns in Neanderthal lineage | [40] |
| 2021 | NGS | Loessal permafrost silts | Analysis of ancient sedimentary DNA from a period of 30,000 years from the central Yukon in Canada. | [14] |
| 2021 | NGS | Mammoth molars | Previous record for the oldest sequenced genome (older than 1 million years). | [25] |
| 2022 | NGS | Sediment | Current record holder for the oldest sequenced DNA | [24] |
| Criterion | Explanation |
|---|---|
| Physically isolated work area | All work on low-copy number DNA should be carried out in an isolated laboratory where no other genetic research is performed. |
| PCR control amplifications | Test laboratory environment for contamination. |
| Test the molecular behavior | Check the PCR products for unusual results. aDNA is heavily fragmented, so longer fragments should be increasingly rarer. |
| Quantitation | Check the number of starting templates. If below 1000, sporadic contamination cannot be ruled out. |
| Reproducibility | Results from the same sample material should be repeatable. |
| Clone | After sequencing, the PCR product should be cloned and sequenced in multiple copies to determine the ratio of exogenous sequences and sequencing errors resulting from aDNA damage. |
| Independent replication | The results should be reproduced in another independent laboratory. |
| Biochemical preservation | Survival of other ancient biomolecules makes the survival of aDNA more believable. |
| Associated remains | If target DNA sequences also survive in associated faunal material, it may be used as supporting evidence. |
| Phylogenetic sense | Reproducible sequences should be placed in a phylogenetic tree with other known haplotypes. |
| Damage patterns | The DNA sequences should show specific damage patterns: a high degree of fragmentation and a high concentration of substitutions on the ends of the fragments (C>T on 5′ and G>A on 3′). |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danielewski, M.; Żuraszek, J.; Zielińska, A.; Herzig, K.-H.; Słomski, R.; Walkowiak, J.; Wielgus, K. Methodological Changes in the Field of Paleogenetics. Genes 2023, 14, 234. https://doi.org/10.3390/genes14010234
Danielewski M, Żuraszek J, Zielińska A, Herzig K-H, Słomski R, Walkowiak J, Wielgus K. Methodological Changes in the Field of Paleogenetics. Genes. 2023; 14(1):234. https://doi.org/10.3390/genes14010234
Chicago/Turabian StyleDanielewski, Mikołaj, Joanna Żuraszek, Aleksandra Zielińska, Karl-Heinz Herzig, Ryszard Słomski, Jarosław Walkowiak, and Karolina Wielgus. 2023. "Methodological Changes in the Field of Paleogenetics" Genes 14, no. 1: 234. https://doi.org/10.3390/genes14010234