
Impact of Data and Study Characteristics on Microbiome Volatility Estimates


Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Measures of Volatility
2.3. Sampling Interval and Sampling Depth Investigations
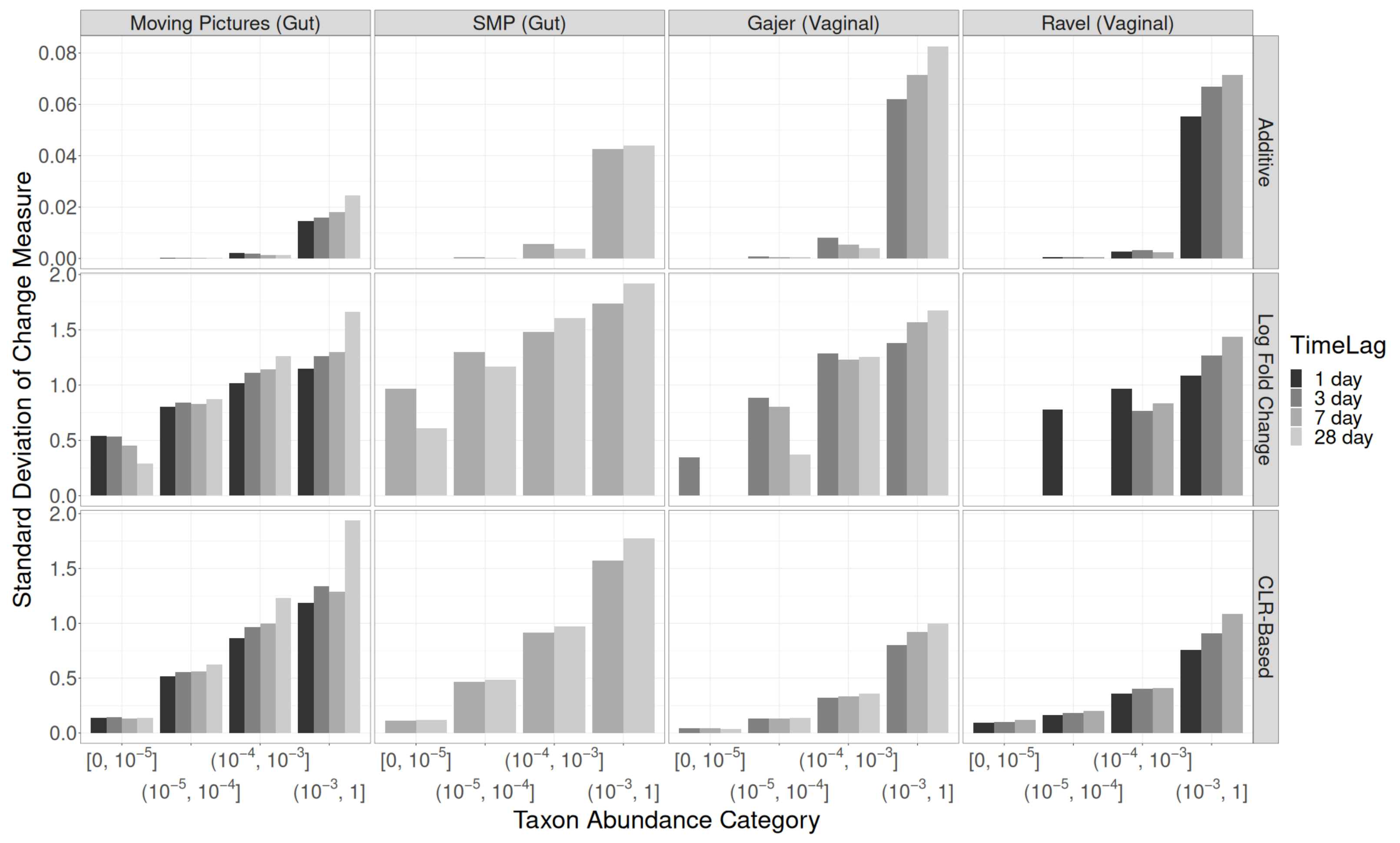
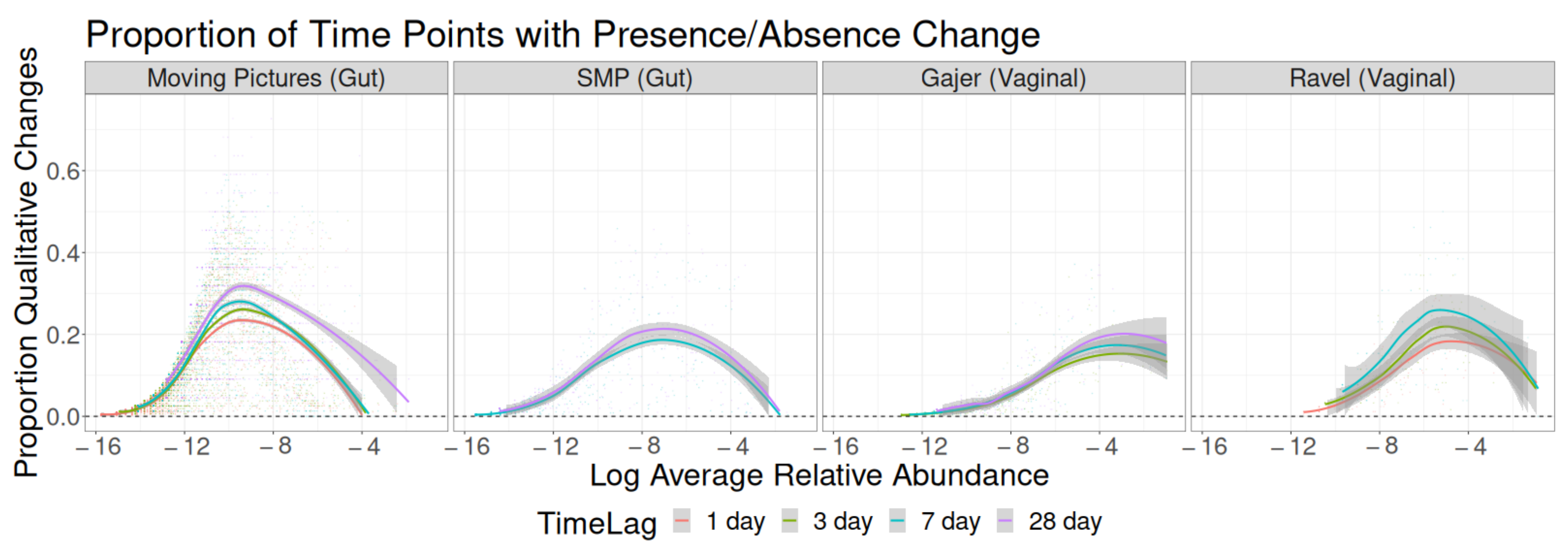
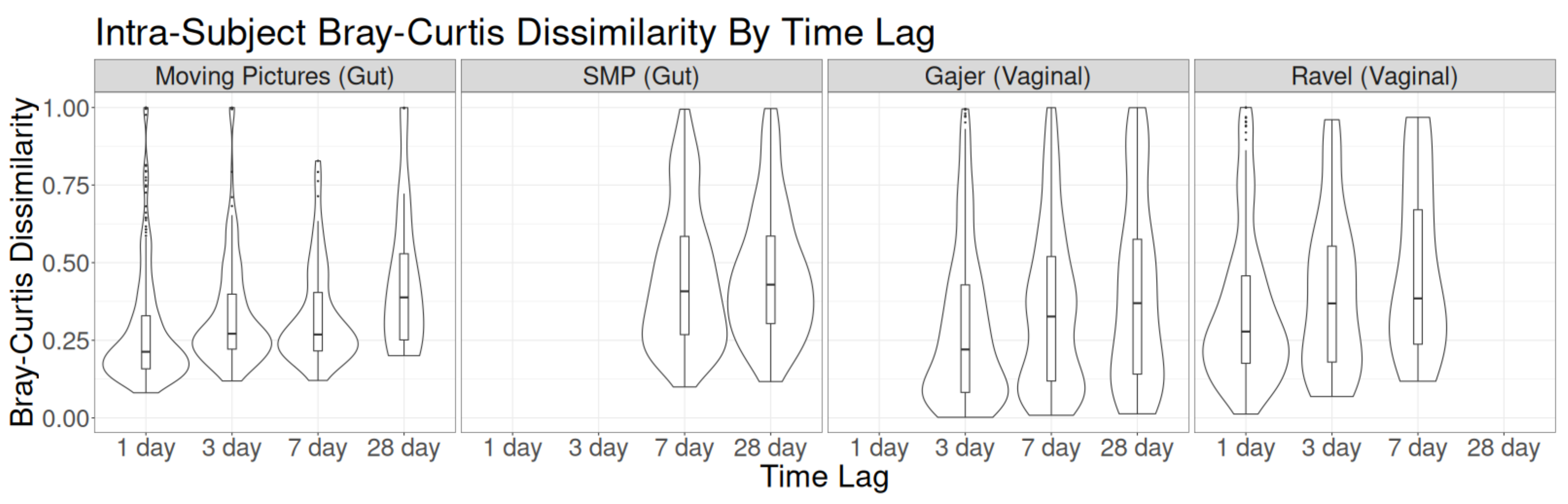
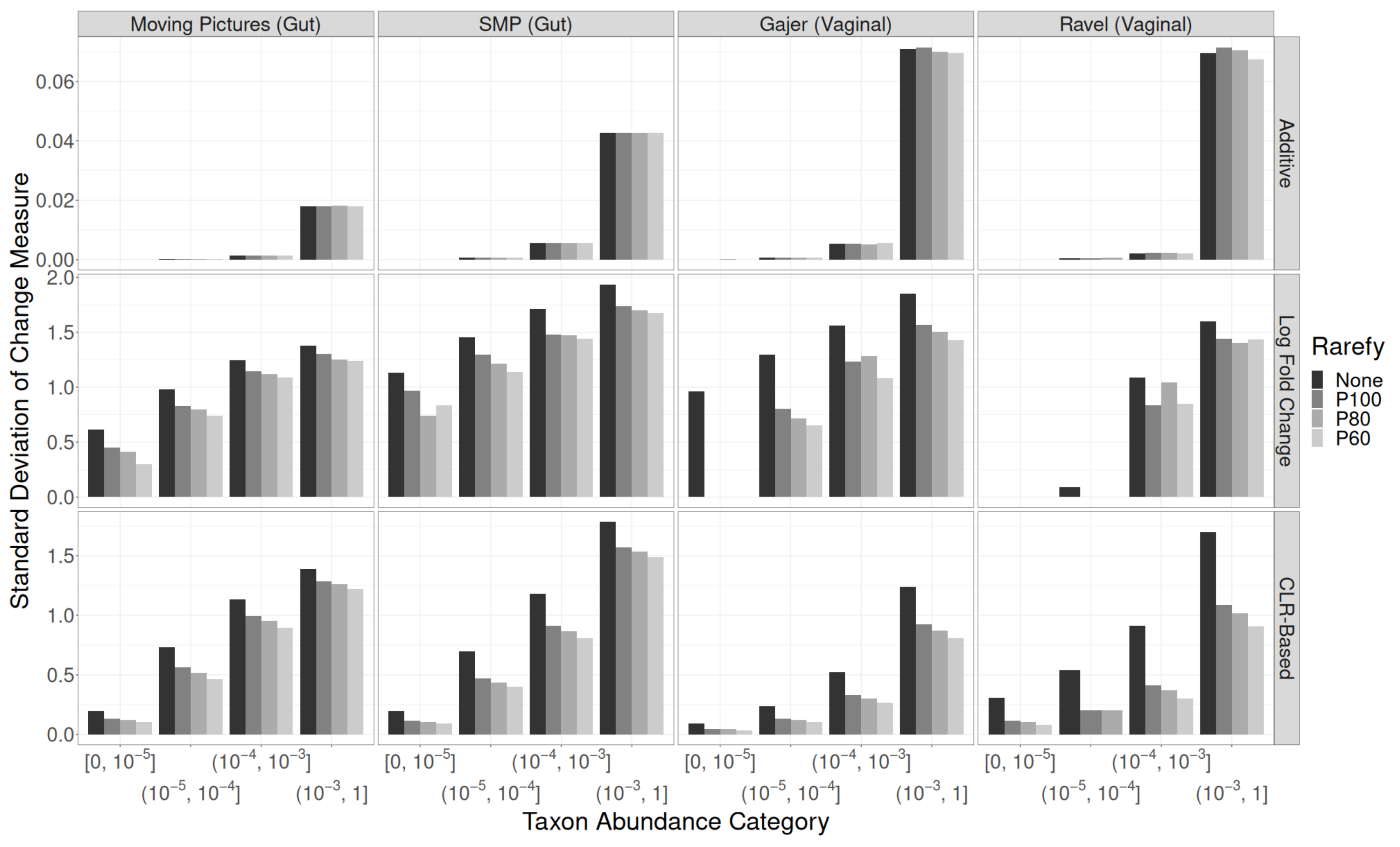
3. Results
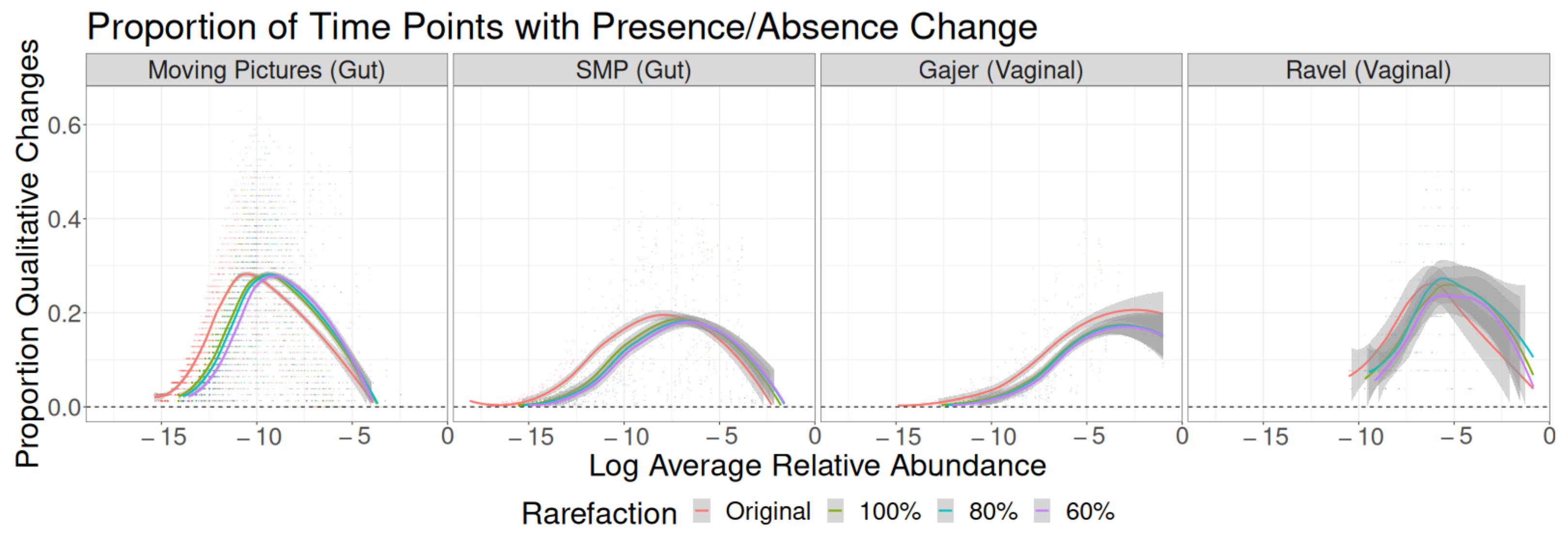
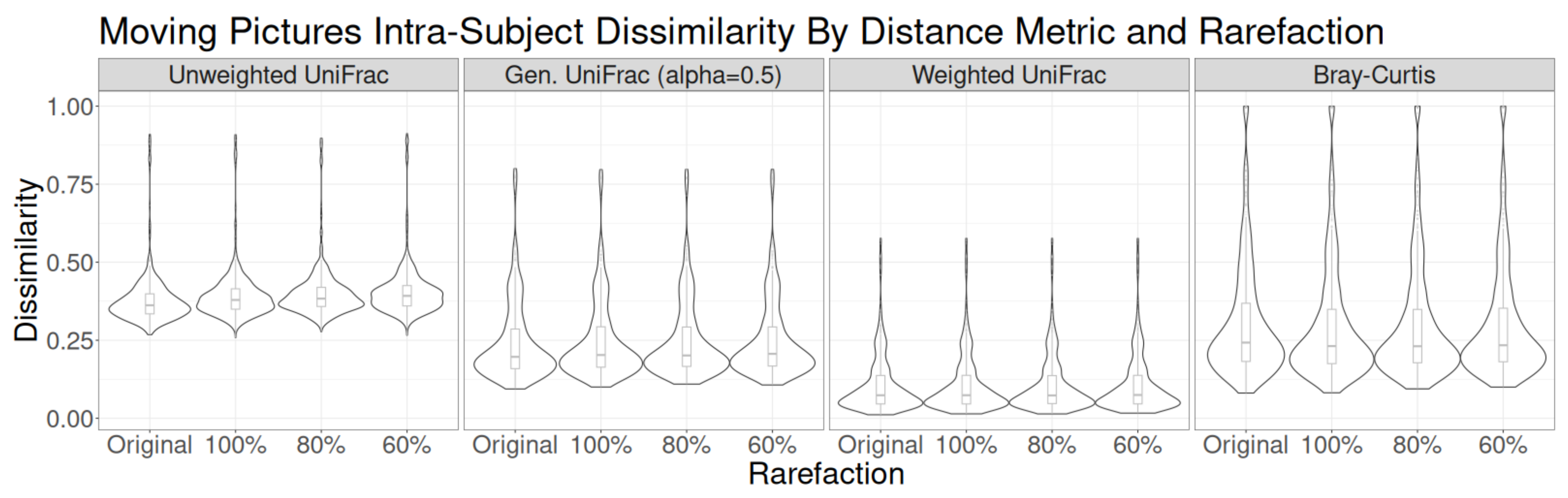
3.1. Sampling Interval Investigations
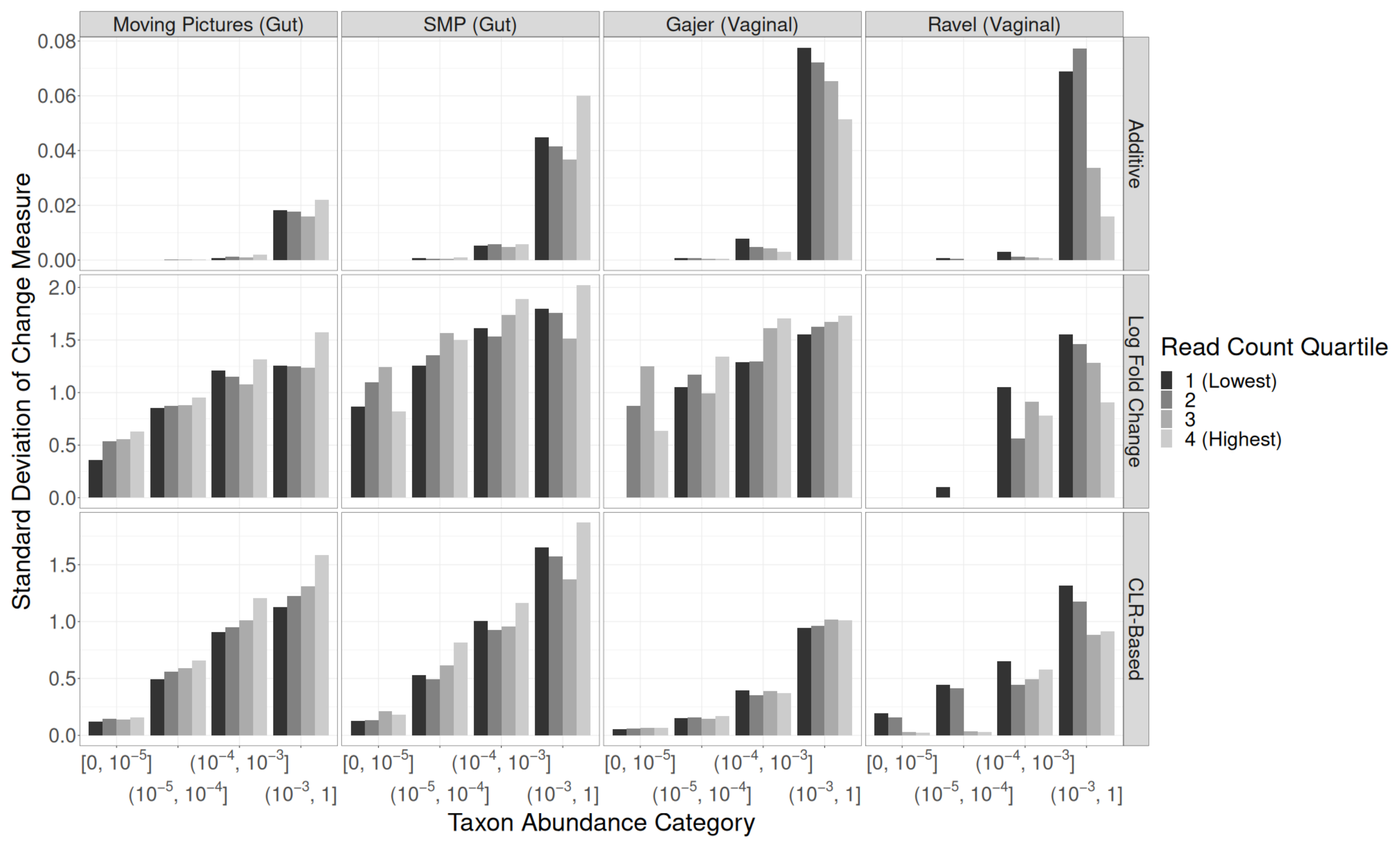
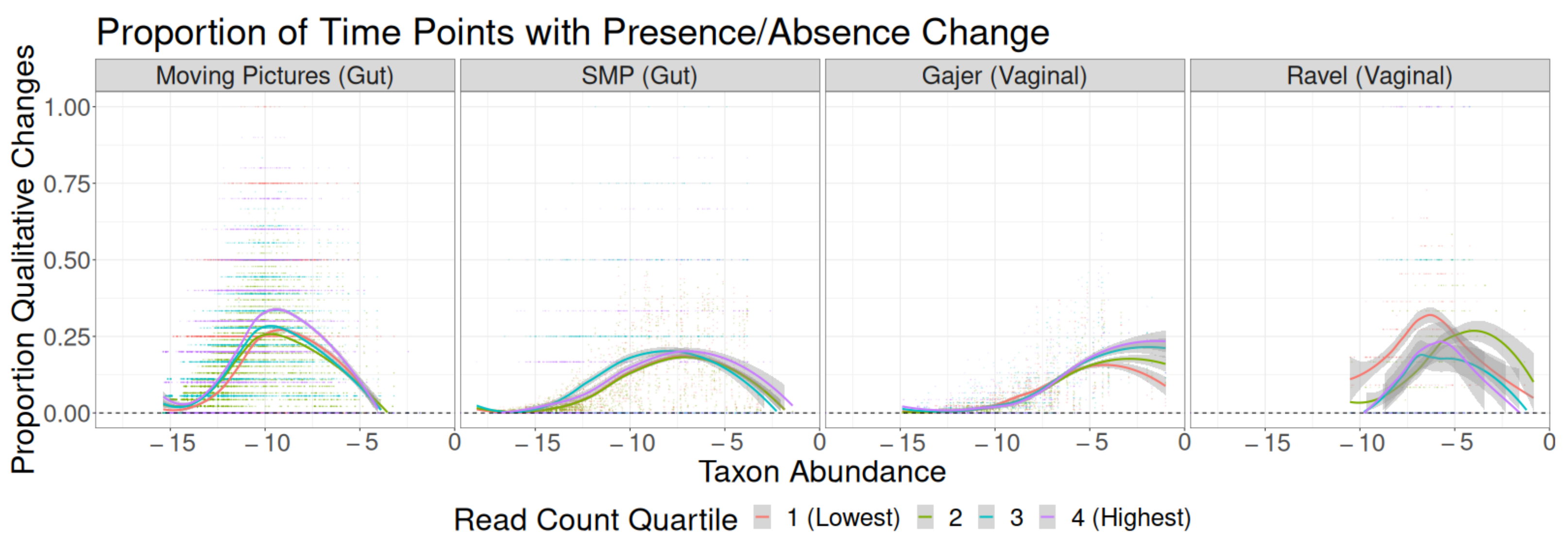
3.2. Read Depth Investigations
Residual Effects of Read Depth after Rarefaction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cox, S.R.; Lindsay, J.O.; Fromentin, S.; Stagg, A.J.; McCarthy, N.E.; Galleron, N.; Ibraim, S.B.; Roume, H.; Levenez, F.; Pons, N.; et al. Effects of low FODMAP diet on symptoms, fecal microbiome, and markers of inflammation in patients with quiescent inflammatory bowel disease in a randomized trial. Gastroenterology 2020, 158, 176–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golob, J.L.; Pergam, S.A.; Srinivasan, S.; Fiedler, T.L.; Liu, C.; Garcia, K.; Mielcarek, M.; Ko, D.; Aker, S.; Marquis, S.; et al. Stool microbiota at neutrophil recovery is predictive for severe acute graft vs host disease after hematopoietic cell transplantation. Clin. Infect. Dis. 2017, 65, 1984–1991. [Google Scholar] [CrossRef] [PubMed]
- Fredricks, D.N.; Plantinga, A.; Srinivasan, S.; Oot, A.; Wiser, A.; Fiedler, T.L.; Proll, S.; Wu, M.C.; Marrazzo, J.M. Vaginal and extra-vaginal bacterial colonization and risk for incident bacterial vaginosis in a population of women who have sex with men. J. Infect. Dis. 2022, 225, 1261–1265. [Google Scholar] [CrossRef] [PubMed]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Costello, E.K.; Berg-Lyons, D.; Gonzalez, A.; Stombaugh, J.; Knights, D.; Gajer, P.; Ravel, J.; Fierer, N.; et al. Moving pictures of the human microbiome. Genome Biol. 2011, 12, R50. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Wang, D.; Garmaeva, S.; Kurilshikov, A.; Vila, A.V.; Gacesa, R.; Sinha, T.; Segal, E.; Weersma, R.K.; Wijmenga, C.; et al. The long-term genetic stability and individual specificity of the human gut microbiome. Cell 2021, 184, 2302–2315. [Google Scholar] [CrossRef]
- Fu, B.C.; Randolph, T.W.; Lim, U.; Monroe, K.R.; Cheng, I.; Wilkens, L.R.; Le Marchand, L.; Lampe, J.W.; Hullar, M.A. Temporal Variability and Stability of the Fecal Microbiome: The Multiethnic Cohort StudyTemporal Variability of the Fecal Microbiome. Cancer Epidemiol. Biomark. Prev. 2019, 28, 154–162. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.J.; Ajami, N.J.; O’Brien, J.L.; Hutchinson, D.S.; Smith, D.P.; Wong, M.C.; Ross, M.C.; Lloyd, R.E.; Doddapaneni, H.; Metcalf, G.A.; et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 2018, 562, 583–588. [Google Scholar] [CrossRef] [Green Version]
- Aho, V.T.; Pereira, P.A.; Voutilainen, S.; Paulin, L.; Pekkonen, E.; Auvinen, P.; Scheperjans, F. Gut microbiota in Parkinson’s disease: Temporal stability and relations to disease progression. EBioMedicine 2019, 44, 691–707. [Google Scholar] [CrossRef] [Green Version]
- Galloway-Peña, J.R.; Smith, D.P.; Sahasrabhojane, P.; Wadsworth, W.D.; Fellman, B.M.; Ajami, N.J.; Shpall, E.J.; Daver, N.; Guindani, M.; Petrosino, J.F.; et al. Characterization of oral and gut microbiome temporal variability in hospitalized cancer patients. Genome Med. 2017, 9, 21. [Google Scholar] [CrossRef]
- Schirmer, M.; Denson, L.; Vlamakis, H.; Franzosa, E.A.; Thomas, S.; Gotman, N.M.; Rufo, P.; Baker, S.S.; Sauer, C.; Markowitz, J.; et al. Compositional and temporal changes in the gut microbiome of pediatric ulcerative colitis patients are linked to disease course. Cell Host Microbe 2018, 24, 600–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.; Karpinets, T.; Prieto, P.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastiaanssen, T.F.; Gururajan, A.; van de Wouw, M.; Moloney, G.M.; Ritz, N.L.; Long-Smith, C.M.; Wiley, N.C.; Murphy, A.B.; Lyte, J.M.; Fouhy, F.; et al. Volatility as a Concept to Understand the Impact of Stress on the Microbiome. Psychoneuroendocrinology 2021, 124, 105047. [Google Scholar] [CrossRef]
- Halfvarson, J.; Brislawn, C.J.; Lamendella, R.; Vázquez-Baeza, Y.; Walters, W.A.; Bramer, L.M.; D’amato, M.; Bonfiglio, F.; McDonald, D.; Gonzalez, A.; et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2017, 2, 17004. [Google Scholar] [CrossRef] [Green Version]
- Plantinga, A.M.; Wu, M.C. Beta Diversity and Distance-Based Analysis of Microbiome Data. In Statistical Analysis of Microbiome Data; Springer: Cham, Switzerland, 2021; pp. 101–127. [Google Scholar]
- Olsson, L.M.; Boulund, F.; Nilsson, S.; Khan, M.T.; Gummesson, A.; Fagerberg, L.; Engstrand, L.; Perkins, R.; Uhlén, M.; Bergström, G.; et al. Dynamics of the normal gut microbiota: A longitudinal one-year population study in Sweden. Cell Host Microbe 2022, 30, 726–739. [Google Scholar] [CrossRef]
- Vandeputte, D.; De Commer, L.; Tito, R.Y.; Kathagen, G.; Sabino, J.; Vermeire, S.; Faust, K.; Raes, J. Temporal variability in quantitative human gut microbiome profiles and implications for clinical research. Nat. Commun. 2021, 12, 6740. [Google Scholar] [CrossRef]
- Mehta, R.S.; Abu-Ali, G.S.; Drew, D.A.; Lloyd-Price, J.; Subramanian, A.; Lochhead, P.; Joshi, A.D.; Ivey, K.L.; Khalili, H.; Brown, G.T.; et al. Stability of the human faecal microbiome in a cohort of adult men. Nat. Microbiol. 2018, 3, 347–355. [Google Scholar] [CrossRef]
- Plantinga, A.M.; Chen, J.; Jenq, R.R.; Wu, M.C. pldist: Ecological dissimilarities for paired and longitudinal microbiome association analysis. Bioinformatics 2019, 35, 3567–3575. [Google Scholar] [CrossRef]
- Silverman, J.D.; Durand, H.K.; Bloom, R.J.; Mukherjee, S.; David, L.A. Dynamic linear models guide design and analysis of microbiota studies within artificial human guts. Microbiome 2018, 6, 202. [Google Scholar] [CrossRef]
- Shenhav, L.; Furman, O.; Briscoe, L.; Thompson, M.; Silverman, J.D.; Mizrahi, I.; Halperin, E. Modeling the temporal dynamics of the gut microbial community in adults and infants. PLoS Comput. Biol. 2019, 15, e1006960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, R.; Goedert, J.J.; Vogtmann, E.; Hua, X.; Porras, C.; Hayes, R.; Safaeian, M.; Yu, G.; Sampson, J.; Ahn, J.; et al. Quantification of human microbiome stability over 6 months: Implications for epidemiologic studies. Am. J. Epidemiol. 2018, 187, 1282–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, G.E.; Caporaso, J.G.; Henley, J.B.; Rideout, J.R.; Domogala, D.; Chase, J.; Leff, J.W.; Vázquez-Baeza, Y.; Gonzalez, A.; Knight, R.; et al. Temporal variability is a personalized feature of the human microbiome. Genome Biol. 2014, 15, 531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajer, P.; Brotman, R.M.; Bai, G.; Sakamoto, J.; Schütte, U.M.; Zhong, X.; Koenig, S.S.; Fu, L.; Ma, Z.; Zhou, X.; et al. Temporal dynamics of the human vaginal microbiota. Sci. Transl. Med. 2012, 4, 132ra52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravel, J.; Brotman, R.M.; Gajer, P.; Ma, B.; Nandy, M.; Fadrosh, D.W.; Sakamoto, J.; Koenig, S.S.; Fu, L.; Zhou, X.; et al. Daily temporal dynamics of vaginal microbiota before, during and after episodes of bacterial vaginosis. Microbiome 2013, 1, 29. [Google Scholar] [CrossRef] [Green Version]
- Martín-Fernández, J.A.; Barceló-Vidal, C.; Pawlowsky-Glahn, V. Dealing with zeros and missing values in compositional data sets using nonparametric imputation. Math. Geol. 2003, 35, 253–278. [Google Scholar] [CrossRef]
- Martín-Fernández, J.A.; Hron, K.; Templ, M.; Filzmoser, P.; Palarea-Albaladejo, J. Model-based replacement of rounded zeros in compositional data: Classical and robust approaches. Comput. Stat. Data Anal. 2012, 56, 2688–2704. [Google Scholar] [CrossRef]
- Martín-Fernández, J.A.; Hron, K.; Templ, M.; Filzmoser, P.; Palarea-Albaladejo, J. Bayesian-multiplicative treatment of count zeros in compositional data sets. Stat. Model. 2015, 15, 134–158. [Google Scholar] [CrossRef]
- Liu, T.; Zhao, H.; Wang, T. An empirical Bayes approach to normalization and differential abundance testing for microbiome data. BMC Bioinform. 2020, 21, 225. [Google Scholar] [CrossRef]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbø, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef]
- Chen, J.; Bittinger, K.; Charlson, E.S.; Hoffmann, C.; Lewis, J.; Wu, G.D.; Collman, R.G.; Bushman, F.D.; Li, H. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics 2012, 28, 2106–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurdie, P.J.; Holmes, S. Waste not, want not: Why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.; Karaoz, U.; de Valpine, P.; Fithian, W. To rarefy or not to rarefy: Robustness and efficiency trade-offs of rarefying microbiome data. Bioinformatics 2022, 38, 2389–2396. [Google Scholar] [CrossRef] [PubMed]
- Cameron, E.S.; Schmidt, P.J.; Tremblay, B.J.M.; Emelko, M.B.; Müller, K.M. To rarefy or not to rarefy: Enhancing diversity analysis of microbial communities through next-generation sequencing and rarefying repeatedly. Sci. Rep. 2021, 11, 22302. [Google Scholar] [CrossRef] [PubMed]
- Willis, A.D. Rarefaction, alpha diversity, and statistics. Front. Microbiol. 2019, 10, 2407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakodi, M.P. Effect of amplicon sequencing depth in environmental microbiome research. Curr. Microbiol. 2021, 78, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Tsilimigras, M.C.; Fodor, A.A. Compositional data analysis of the microbiome: Fundamentals, tools, and challenges. Ann. Epidemiol. 2016, 26, 330–335. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Caporaso et al., 2011 [5] | Flores et al., 2014 [24] | Gajer et al., 2012 [25] | Ravel et al., 2013 [26] | |
|---|---|---|---|---|
| Basic Study Information | ||||
| Study name | Moving Pictures | SMP | - | - |
| Body site | Gut | Gut | Vagina | Vagina |
| Number of subjects | 2 | 58 | 32 | 6 |
| Percent female | 50% | 63.7% | 100% | 100% |
| Percent white | - | 75.9% | 40.6% | 16.7% |
| Age (years): Mean (SD) | - | 24.1 (6.4) | 37.1 (8.1) | 27.2 (6.3) |
| Age (years): Range | 32–33 | 18–55 | 22–53 | 21–38 |
| Sampling Frequency and Study Duration | ||||
| Number of time points | 131–336 | 7–10 | 25–33 | 23–38 |
| Sampling interval | Daily | Weekly | Twice-weekly | Daily |
| Study duration | 6–15 months | 3 months | 16 weeks | 10 weeks |
| Summaries of Taxa and Reads | ||||
| Read count: Median | 36,114 | 43,282 | 2403 | 5195 |
| Read count: Range | 15,355–60,847 | 11,393–188,192 | 556–6619 | 145–15,972 |
| Number of unique taxa | 3962 | 632 | 331 | 122 |
| Taxon analysis level | Genus | Genus | Genus/Species | Species |
| Definition | Requirements | Possible Values | |
|---|---|---|---|
| Additive | - | ||
| Multiplicative | |||
| CLR-Based | computed after zero-replacement | ||
| Qualitative | - | −1 (present → absent), 0, | |
| 1 (absent → present) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, D.J.; Plantinga, A.M. Impact of Data and Study Characteristics on Microbiome Volatility Estimates. Genes 2023, 14, 218. https://doi.org/10.3390/genes14010218
Park DJ, Plantinga AM. Impact of Data and Study Characteristics on Microbiome Volatility Estimates. Genes. 2023; 14(1):218. https://doi.org/10.3390/genes14010218
Chicago/Turabian StylePark, Daniel J., and Anna M. Plantinga. 2023. "Impact of Data and Study Characteristics on Microbiome Volatility Estimates" Genes 14, no. 1: 218. https://doi.org/10.3390/genes14010218