
Weighted Gene Co-Expression Network Analysis Reveals Hub Genes for Fuzz Development in Gossypium hirsutum

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. RNA Extraction, Library Construction, and Sequencing
2.3. RNA-Seq Data Analysis
2.4. Construction of Weighted Gene Co-Expression Network
2.5. Screening of Specific Modules Associated with Fuzz
2.6. Enrichment Analysis
2.7. qRT-PCR Validated Transcriptome Sequencing
3. Results and Analysis
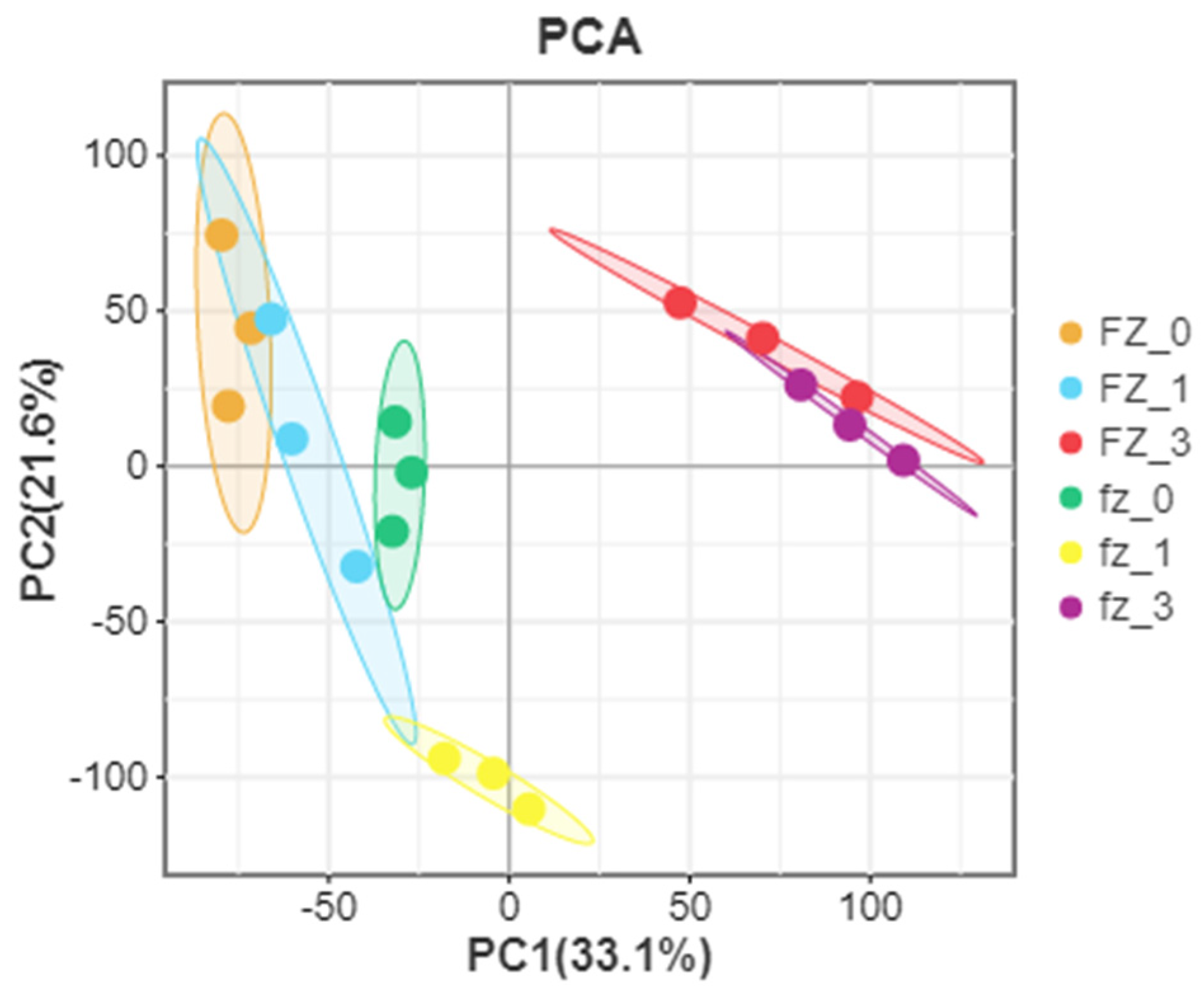
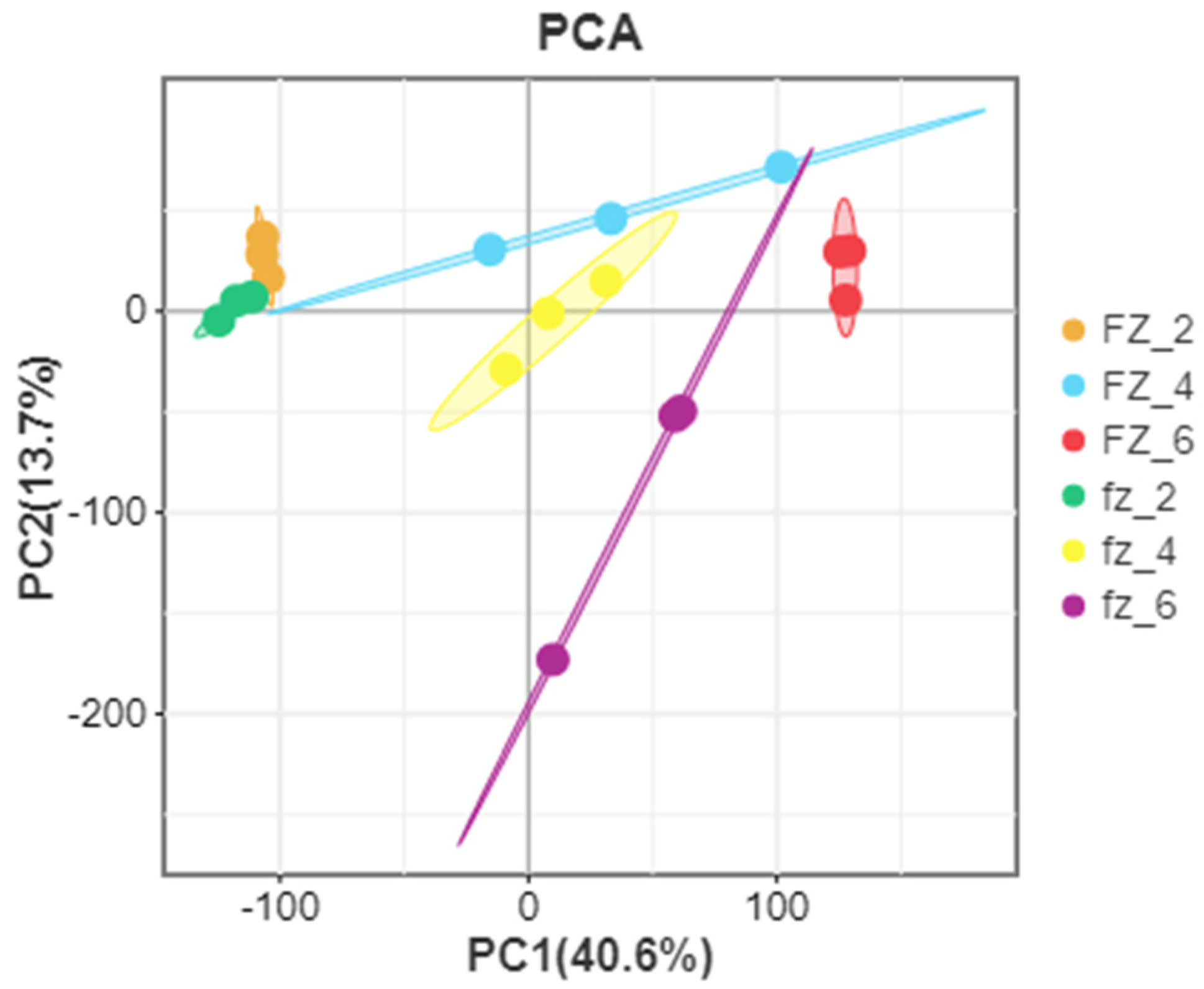
3.1. Data Processing and Analysis
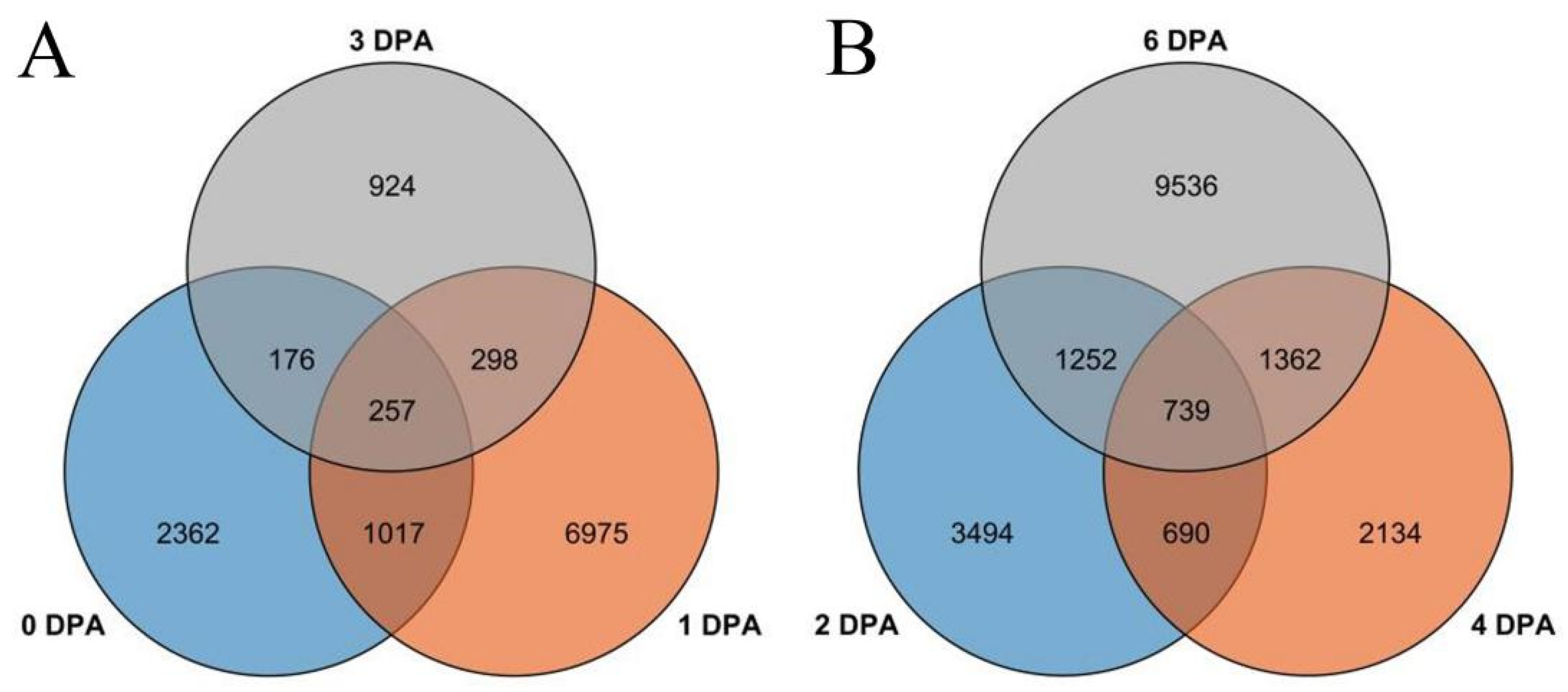
3.2. Differential Gene Expression Analysis of FZ and fz
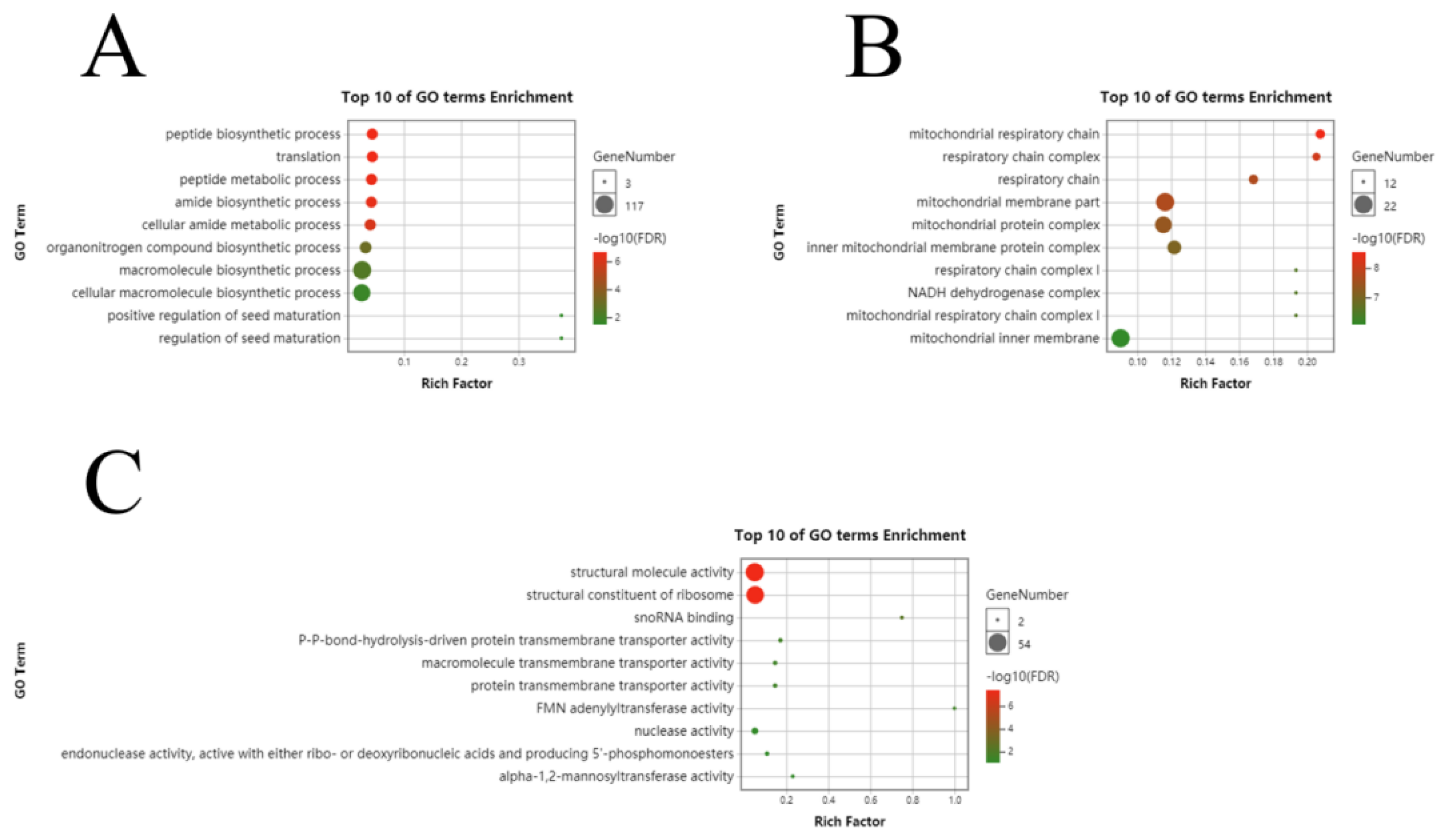
3.3. Functional Analysis of Differential Genes
3.4. Consistency Evaluation of qRT-PCR and RNA-Seq
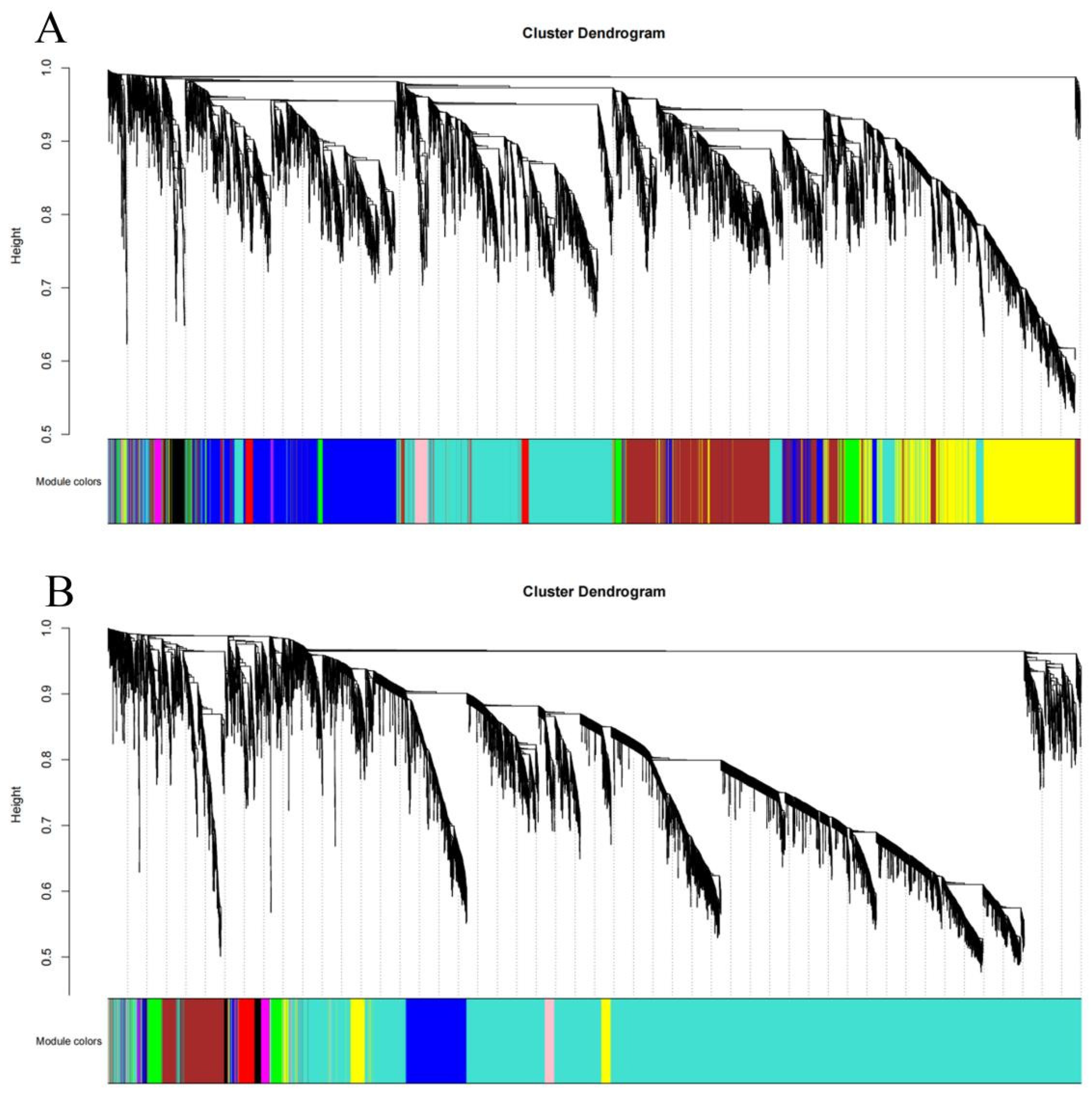
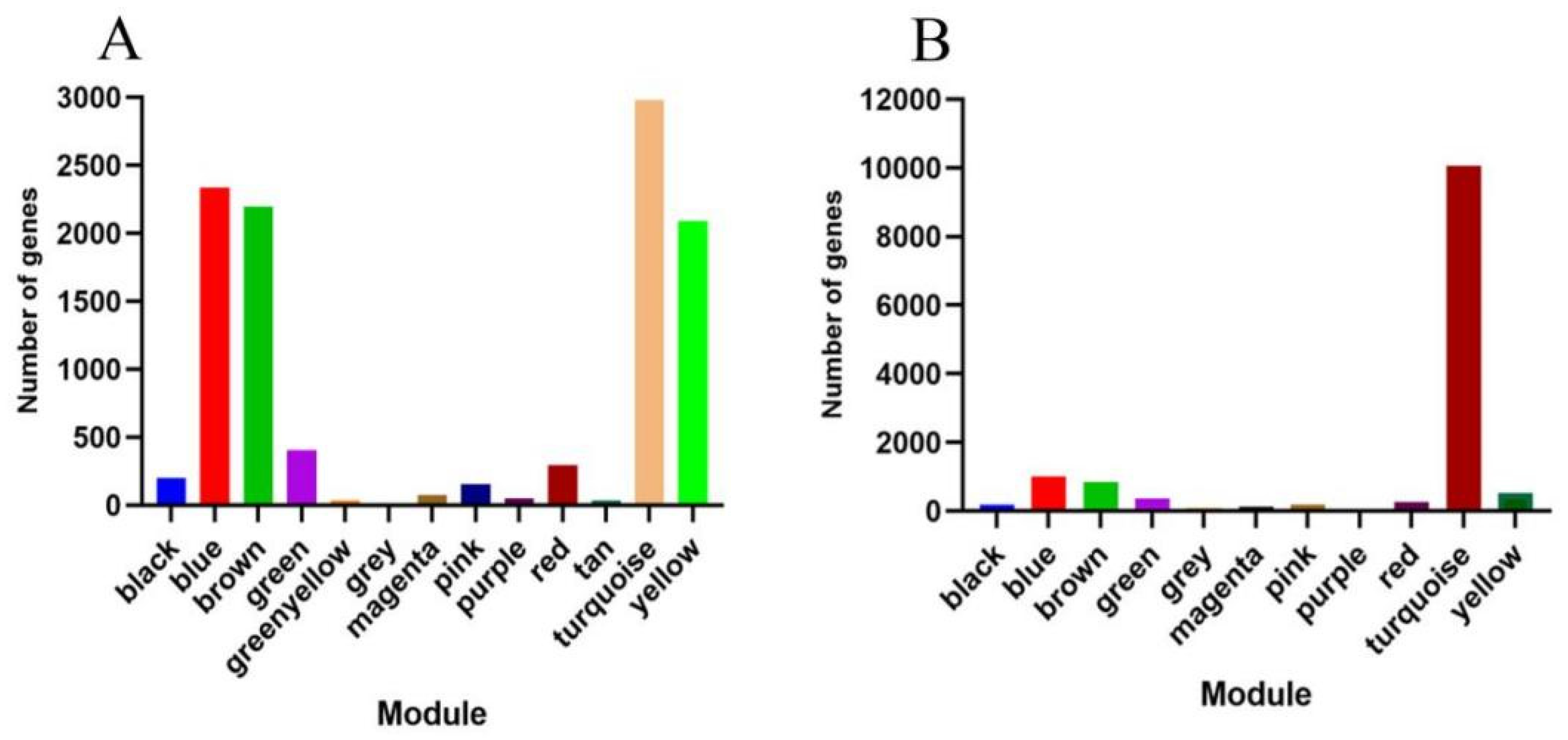
3.5. Construction of Weighted Gene Co-Expression Network
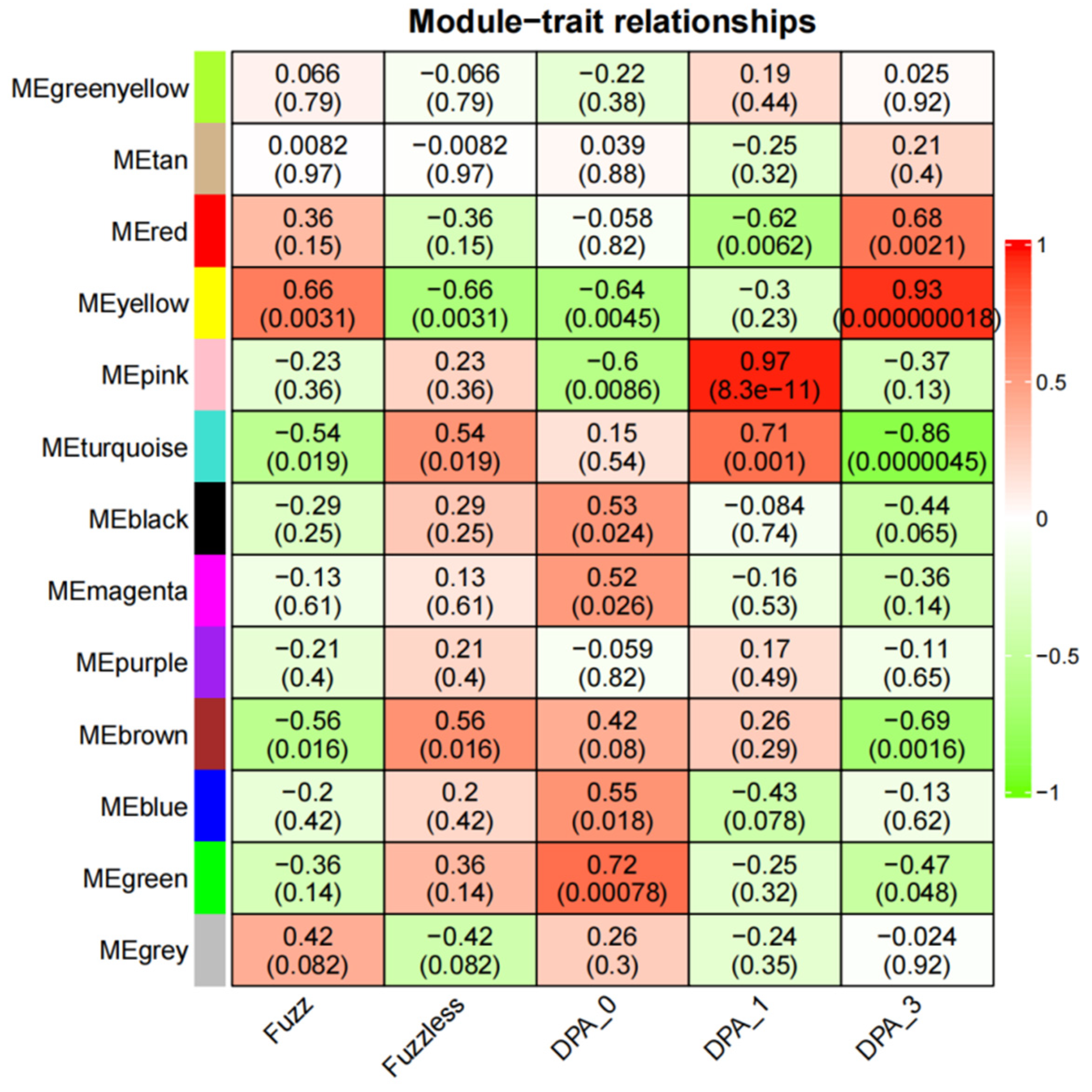
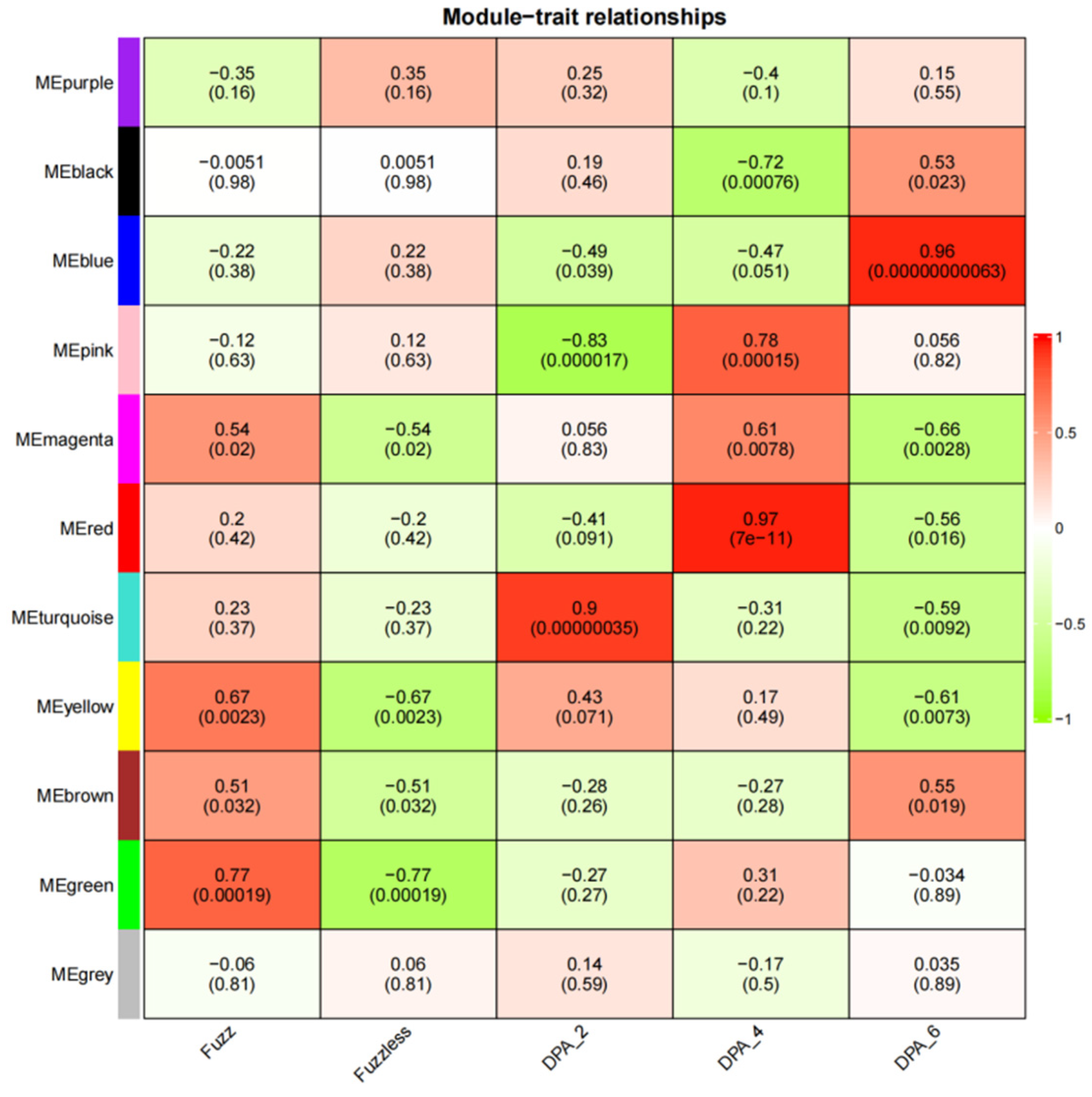
3.6. Identification of Specific Modules Related to Fuzz
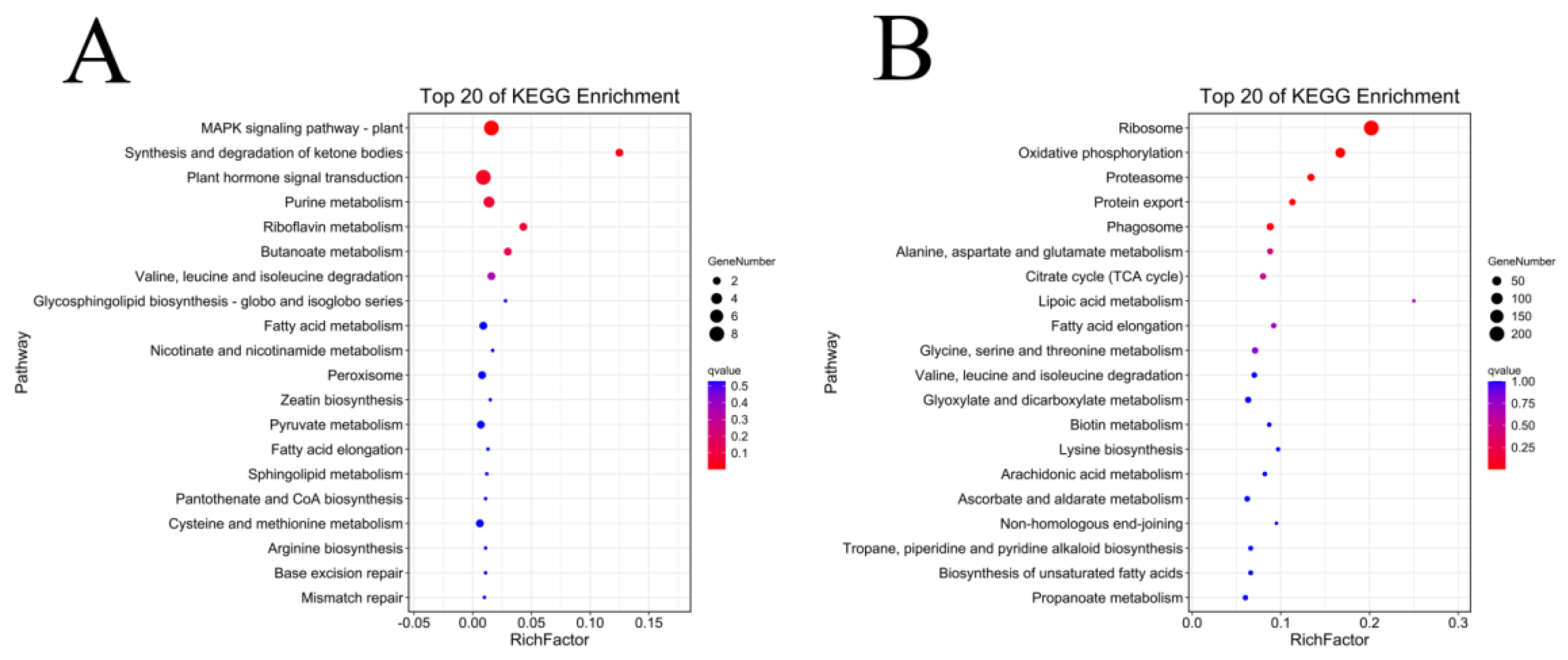
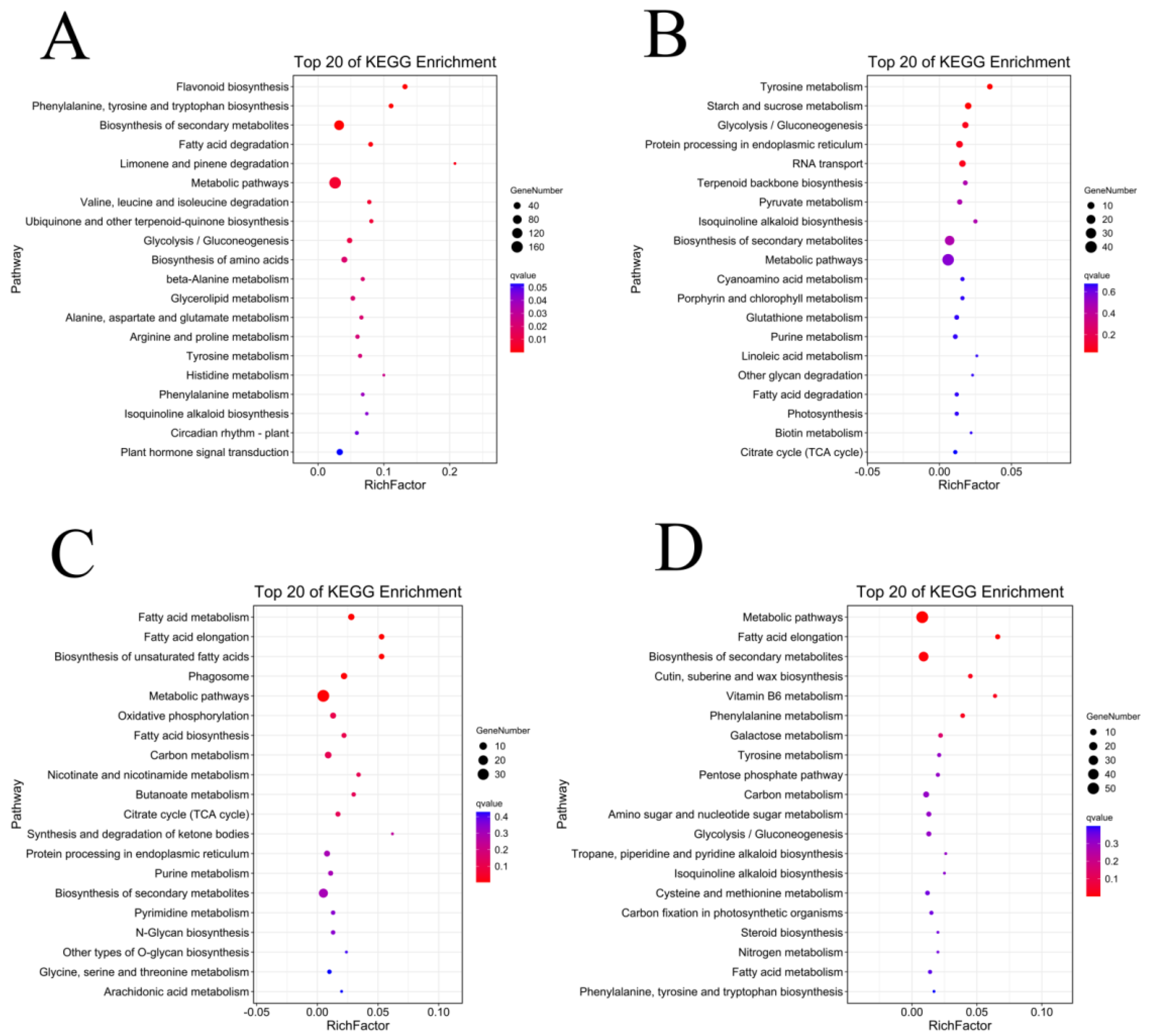
3.7. Functional Enrichment Analysis of Development-Specific Modules with Fuzz
3.8. Analysis of Hub Genes of Modules Related to Fuzz Development in Four Materials
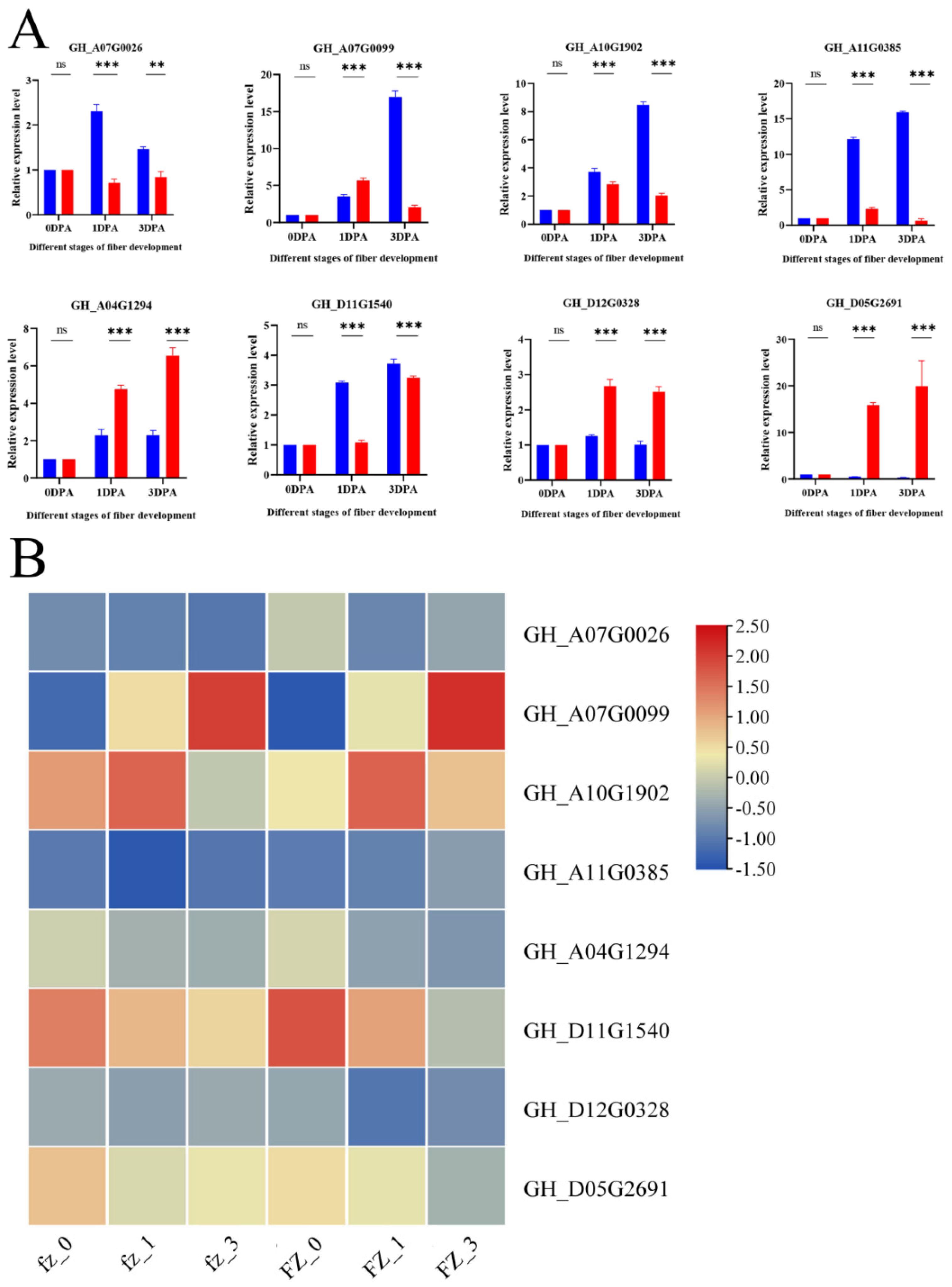
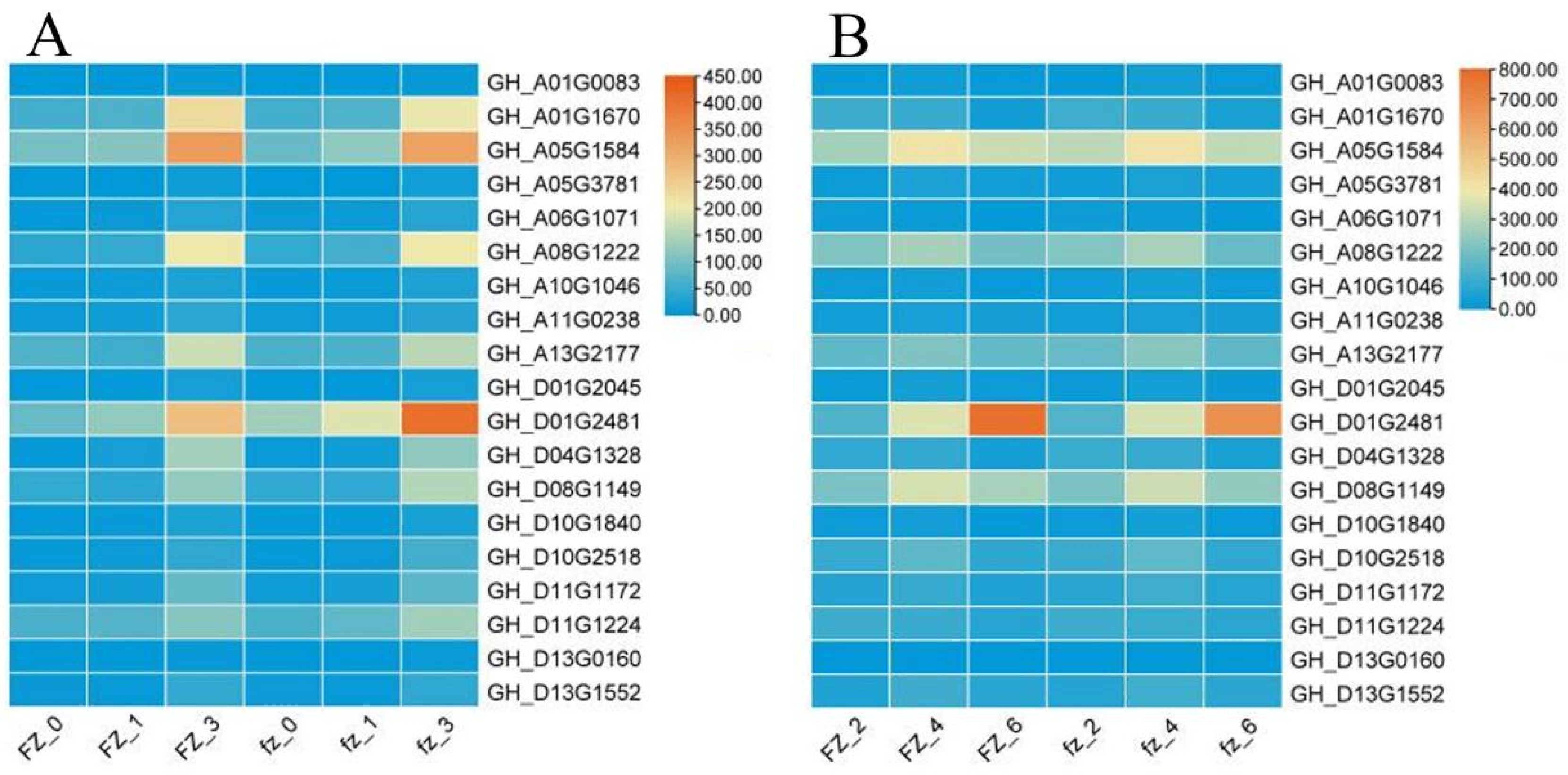
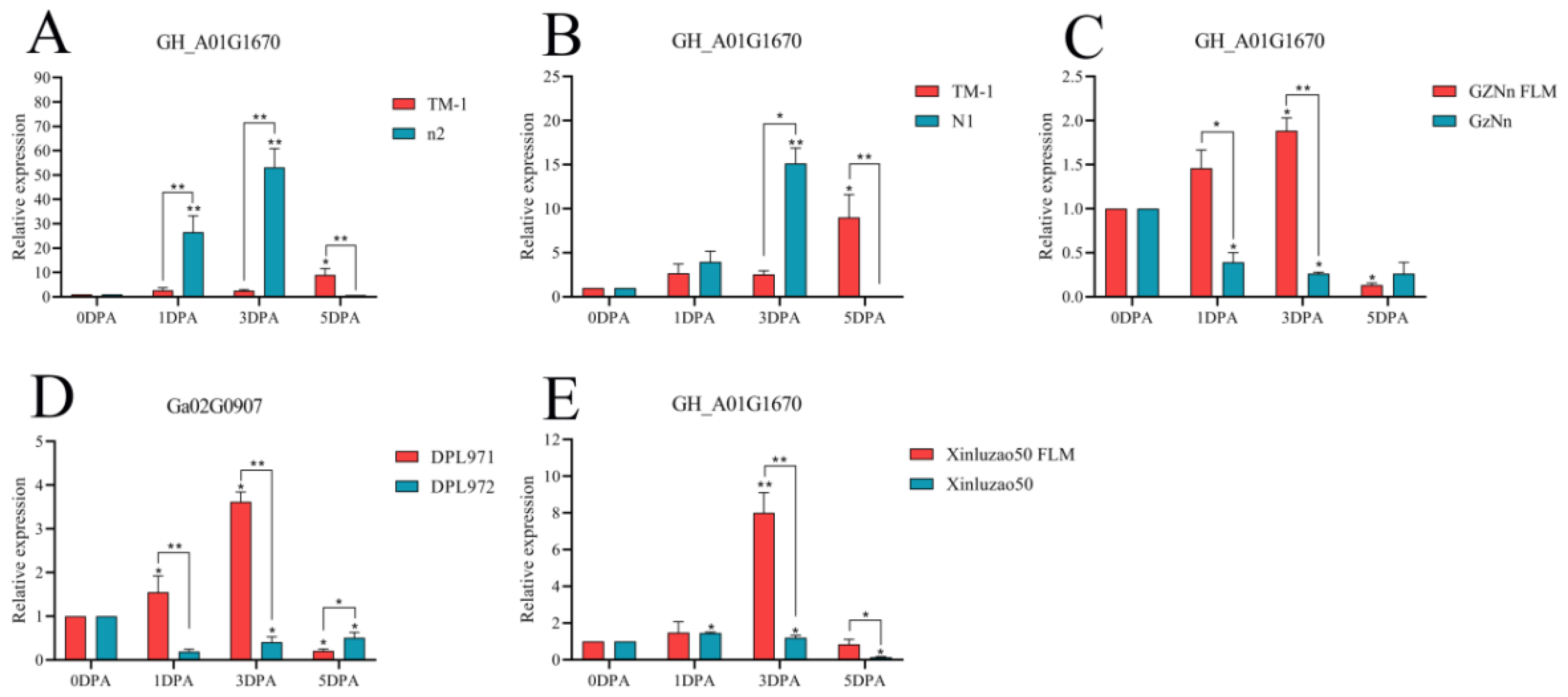
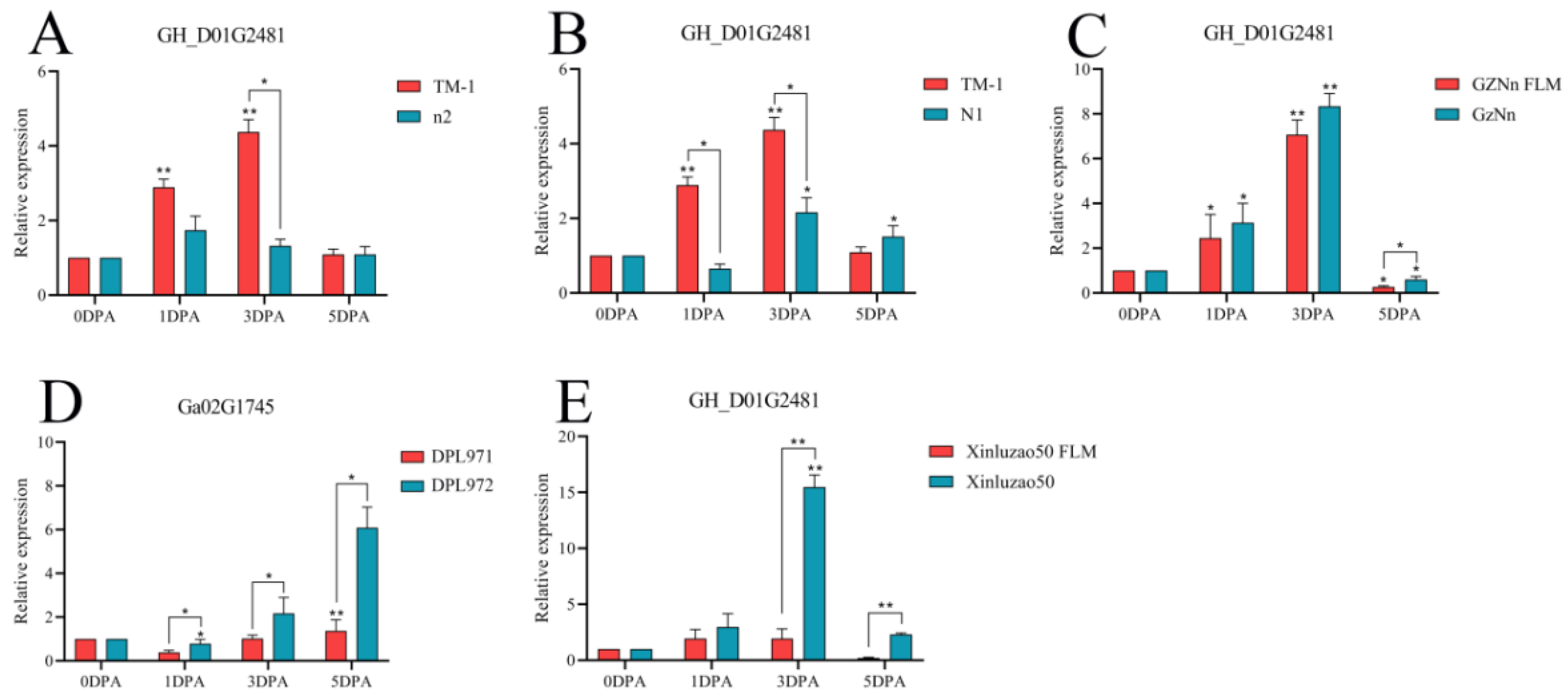
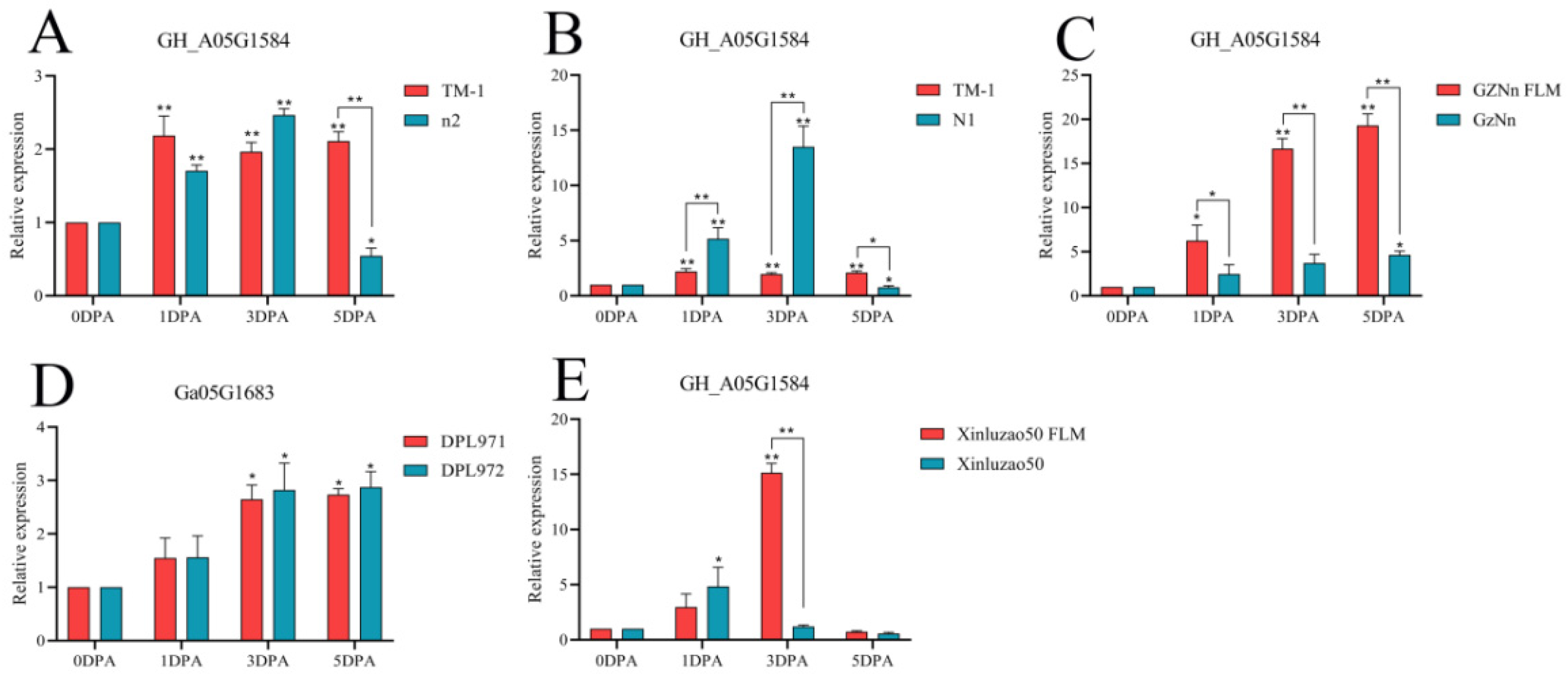
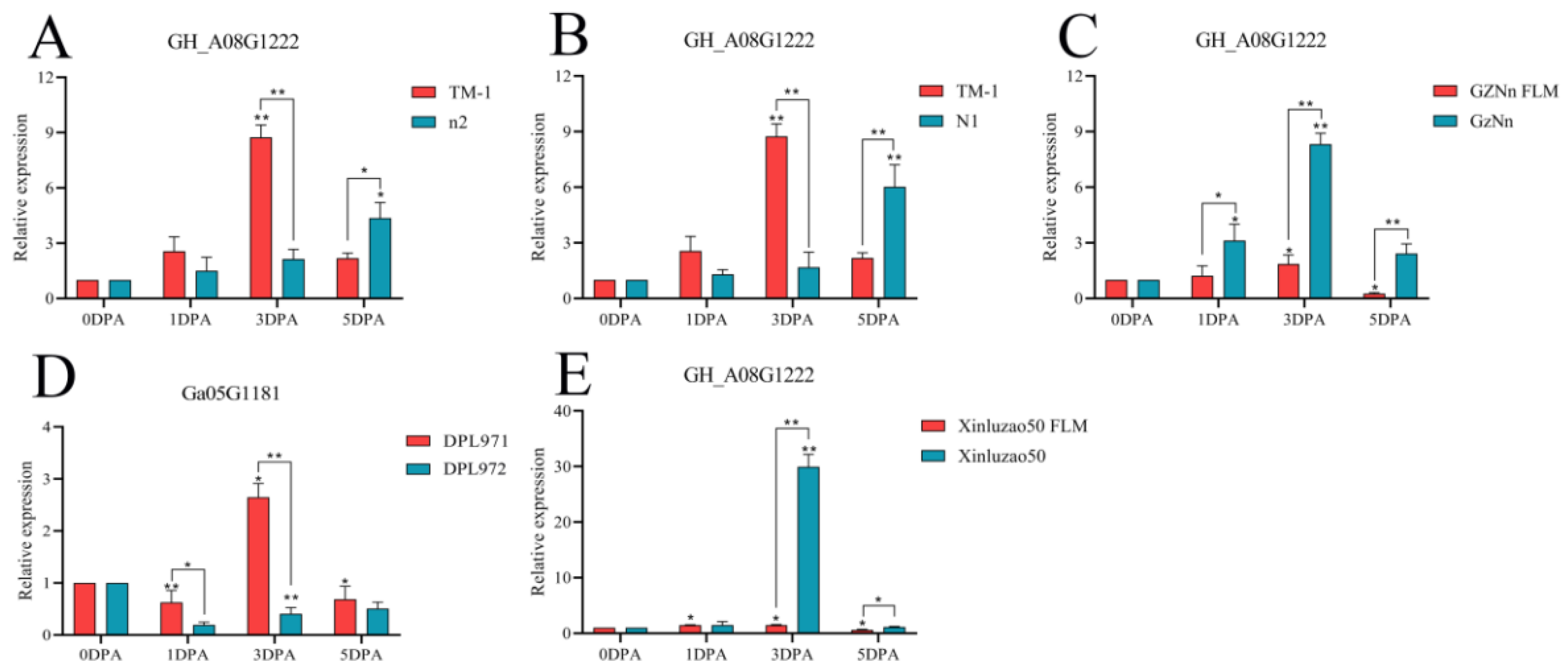
3.9. qRT-PCR of Hub Genes
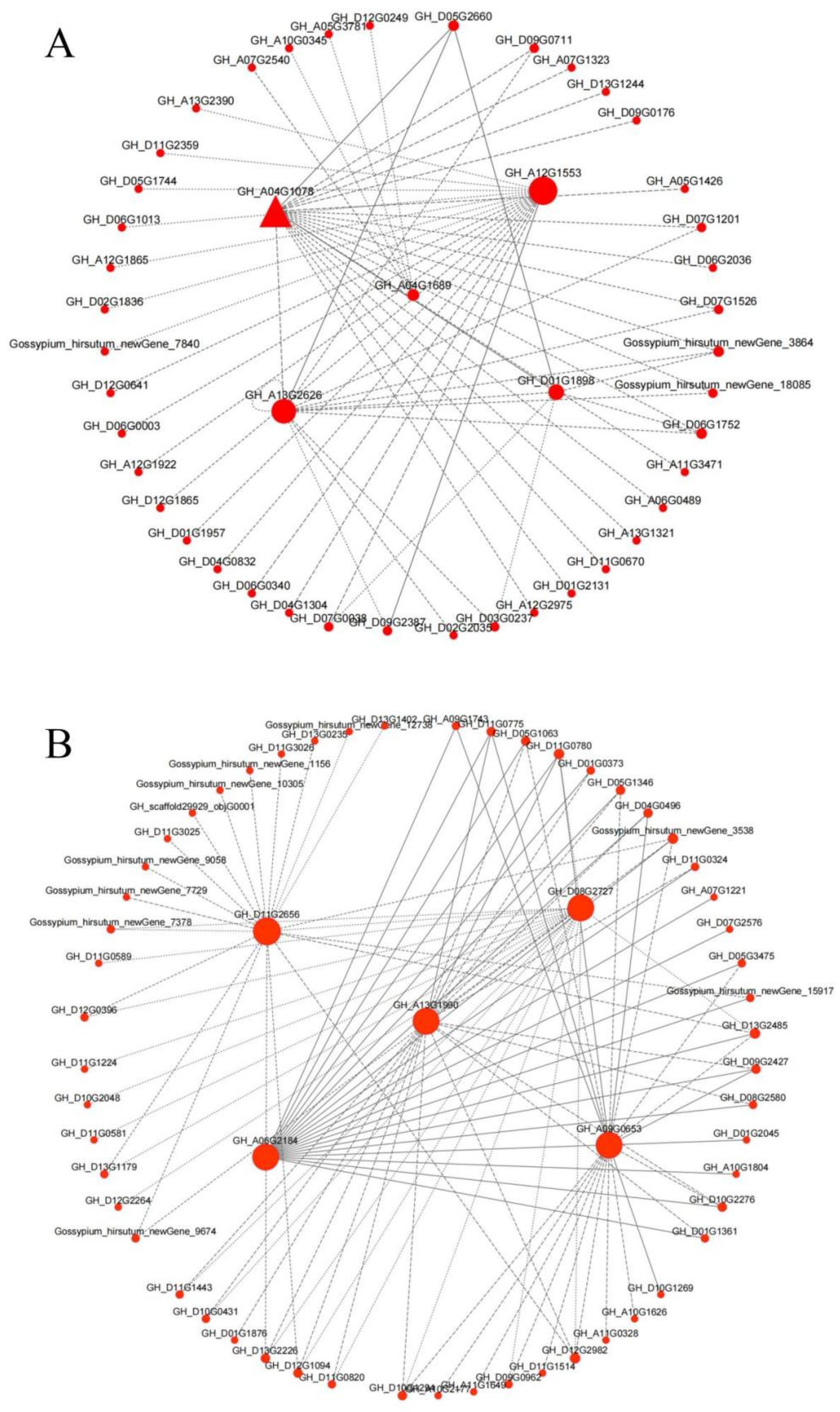
3.10. Module Hub Gene Mining and Interaction Network Analysis
4. Discussion
4.1. Hub Genes in the Six Modules May Be Involved in the Formation of Cotton Fuzz
4.2. Some Genes Are Involved in Fuzz Development of Two Kinds of Fuzzless Upland Cotton
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, J.J.; Woodward, A.W.; Chen, Z.J. Gene expression changes and early events in cotton fibre development. Ann. Bot. 2007, 100, 1391–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterson, A.H.; Wendel, J.F.; Gundlach, H.; Guo, H.; Jenkins, J.; Jin, D.; Llewellyn, D.; Showmaker, K.C.; Shu, S.; Udall, J.; et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 2012, 492, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Zhou, L.; Zhang, W.; Cai, L.; Guo, H.; Tian, H.; Schiefelbein, J.; Wang, S. A single amino acid substitution in the R3 domain of GLABRA1 leads to inhibition of trichome formation in Arabidopsis without affecting its interaction with GLABRA3. Plant Cell Environ. 2015, 39, 897–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.; Walford, S.A.; Dennis, E.S.; Llewellyn, D.J. Trichomes at the Base of the Petal Are Regulated by the Same Transcription Factors as Cotton Seed Fibers. Plant Cell Physiol. 2020, 61, 1590–1599. [Google Scholar] [CrossRef]
- Matias-Hernandez, L.; Aguilar-Jaramillo, A.E.; Cigliano, R.A.; Sanseverino, W.; Pelaz, S. Flowering and trichome development share hormonal and transcription factor regulation. J. Exp. Bot. 2016, 67, 1209–1219. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Ye, Z. Trichomes as models for studying plant cell differentiation. Cell Mol. Life Sci. 2013, 70, 1937–1948. [Google Scholar] [CrossRef]
- Pattanaik, S.; Patra, B.; Singh, S.K.; Yuan, L. An overview of the gene regulatory network controlling trichome development in the model plant, Arabidopsis. Front. Plant Sci. 2014, 5, 259. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Sano, R.; Wada, T.; Takabayashi, J.; Okada, K. Jasmonic acid control of GLABRA3 links inducible defense and trichome patterning in Arabidopsis. Development 2009, 136, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Gao, D.; Xiong, Y.; Tang, X.; Xiao, X.; Wang, C.; Yu, S. Hairy Leaf 6, an AP2/ERF transcription factor, interacts with OsWOX3B and regulates trichome formation in rice. Mol. Plant 2017, 10, 1417–1433. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wu, K.; Wang, Y.; Peng, Y.; Hu, F.; Wen, L.; Han, B.; Qian, Q.; Teng, S. A WUSCHEL-like homeobox gene, OsWOX3B responses to NUDA/GL-1 locus in rice. Rice J. 2012, 5, 30. [Google Scholar] [CrossRef]
- Vernoud, V.; Laigle, G.; Rozier, F.; Meeley, R.B.; Perez, P.; Rogowsky, P.M. The HD-ZIP IV transcription factor OCL4 is necessary for trichome patterning and anther development in maize. Plant J. Cell Mol. Biol. 2009, 59, 883–894. [Google Scholar] [CrossRef]
- Kong, D.; Pan, X.; Jing, Y.; Zhao, Y.; Duan, Y.; Yang, J.; Wang, B.; Liu, Y.; Shen, R.; Cao, Y.; et al. ZmSPL10/14/26 are required for epidermal hair cell fate specification on maize leaf. New Phytol. 2021, 230, 1533–1549. [Google Scholar] [CrossRef]
- Xu, J.; van Herwijnen, Z.O.; Drager, D.B.; Sui, C.; Haring, M.A.; Schuurink, R.C. SlMYC1 Regulates Type VI Glandular Trichome Formation and Terpene Biosynthesis in Tomato Glandular Cells. Plant Cell 2018, 30, 2988–3005. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Li, H.; Zhang, J.; Luo, Z.; Gong, P.; Zhang, C.; Li, J.; Wang, T.; Zhang, Y.; Lu, Y.; et al. A regulatory gene induces trichome formation and embryo lethality in tomato. Proc. Natl. Acad. Sci. USA 2011, 108, 11836–11841. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.K.D.; Ruan, Y. Looking into ‘hair tonics’ for cotton fiber initiation. New Phytol. 2020, 229, 1844–1851. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wu, S.; Nowak, J.; Wang, G.; Han, L.; Feng, Z.; Mendrinna, A.; Ma, Y.; Wang, H.; Zhang, X.; et al. Live-cell imaging of the cytoskeleton in elongating cotton fibres. Nat. Plants 2019, 5, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, G.; Long, L.; Altunok, S.; Feng, Z.; Wang, D.; Khawar, K.M.; Mujtaba, M. Understanding the role of phytohormones in cotton fiber development through omic approaches; recent advances and future directions. Int. J. Biol. Macromol. 2020, 163, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Mei, G.; Zhang, Z. Optimization of polar distribution of GhPIN3a in the ovule epidermis improves cotton fiber development. J. Exp. Bot. 2019, 70, 3021–3023. [Google Scholar] [CrossRef]
- Zeng, J.; Zhang, M.; Hou, L.; Bai, W.; Yan, X.; Hou, N.; Wang, H.; Huang, J.; Zhao, J.; Pei, Y. Cytokinin inhibits cotton fiber initiation by disrupting PIN3a-mediated asymmetric accumulation of auxin in the ovule epidermis. J. Exp. Bot. 2019, 70, 3139–3151. [Google Scholar] [CrossRef] [Green Version]
- Xiao, G.; He, P.; Zhao, P.; Liu, H.; Zhang, L.; Pang, C.; Yu, J. Genome-wide identification of the GhARF gene family reveals that GhARF2 and GhARF18 are involved in cotton fibre cell initiation. J. Exp. Bot. 2018, 69, 4323–4337. [Google Scholar] [CrossRef]
- Zhang, M.; Zeng, J.Y.; Long, H.; Xiao, Y.H.; Yan, X.Y.; Pei, Y. Auxin Regulates Cotton Fiber Initiation via GhPIN-Mediated Auxin Transport. Plant Cell Physiol. 2017, 58, 385–397. [Google Scholar] [CrossRef]
- Walford, S.A.; Wu, Y.; Llewellyn, D.J.; Dennis, E.S. Epidermal cell differentiation in cotton mediated by the homeodomain leucine zipper gene, GhHD-1. Plant J. Cell Mol. Biol. 2012, 71, 464–478. [Google Scholar] [CrossRef]
- Shangguan, X.X.; Yang, C.Q.; Zhang, X.F.; Wang, L.J. Functional characterization of a basic helix-loop-helix (bHLH) transcription factor GhDEL65 from cotton (Gossypium hirsutum). Physiol. Plant 2016, 158, 200–212. [Google Scholar] [CrossRef]
- Xu, Y.; Shi, Y.; Feng, J.; Zhu, Y. Detection of gene expression related to cotton fiber development by cDNA chip. Mol. Plant Breed. 2004, 3, 348–353. [Google Scholar]
- Wang, M.J.; Tu, L.L.; Yuan, D.J.; Zhu, D.; Shen, C.; Li, J.Y.; Liu, F.Y.; Pei, L.L.; Wang, P.C.; Zhao, G.N.; et al. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 2018, 51, 224–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Huang, G.; He, S.; Yang, Z.; Sun, G.; Ma, X.; Li, N.; Zhang, X.; Sun, J.; Liu, M.; et al. Resequencing of 243 diploid cotton accessions based on an updated a genome identifies the genetic basis of key agronomic traits. Nat. Genet. 2018, 50, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.S.; Fang, D.D.; Thyssen, G.N.; Delhom, C.D.; Liu, Y.L.; Kim, H.J. Comparative fiber property and transcriptome analyses reveal key genes potentially related to high fiber strength in cotton (Gossypium hirsutum L.) line MD52ne. BMC Plant Biol. 2016, 16, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiaoyang, W.; Liyuan, W.; Zhaoe, P.; Shoupu, H.; Xiao, W.; Wenfang, G.; Xiongming, D. Fiber development and differential gene expression analysis of Asian cotton linters mutants. J. Crops 2020, 46, 645–660. [Google Scholar]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Liu, S.; Song, G. Weighted Gene Co-Expression Network Analysis Reveals Hub Genes Contributing to Fuzz Development in Gossypium arboreum. Genes 2021, 12, 753. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Fan, L.; Li, P. Co-expression network and comparative transcriptome analysis for fiber initiation and elongation reveal genetic differences in two lines from upland cotton CCRI70 RIL population. PeerJ 2021, 9, e11812. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Zhang, T. MIXTAs and phytohormones orchestrate cotton fiber development. Curr. Opin. Plant Biol. 2021, 59, 101975. [Google Scholar] [CrossRef]
- Wan, Q.; Guan, X.; Yang, N.; Wu, H.; Pan, M.; Liu, B.; Fang, L.; Yang, S.; Hu, Y.; Ye, W.; et al. Small interfering RNAs from bidirectional transcripts of GhMML3_A12 regulate cotton fiber development. New Phytol. 2016, 210, 1298–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Li, Y.; Zhu, S.; Fang, S.; Zhao, L.; Guo, Y.; Wang, J.; Yuan, L.; Lu, Y.; Liu, F.; et al. A Retrotransposon Insertion in GhMML3_D12 Is Likely Responsible for the Lintless Locus li3 of Tetraploid Cotton. Front. Plant Sci. 2020, 11, 593679. [Google Scholar] [CrossRef]
- Wu, H.; Tian, Y.; Wan, Q.; Fang, L.; Guan, X.; Chen, J.; Hu, Y.; Ye, W.; Zhang, H.; Guo, W.; et al. Genetics and evolution of MIXTA genes regulating cotton lint fiber development. New Phytol. 2017, 217, 883–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.-H.; Yuan, Y.; Stiller, W.; Jia, Y.; Wang, P.; Pan, Z.; Du, X.; Llewellyn, D.; Wilson, I. Genetic dissection of the fuzzless seed trait in Gossypium barbadense. J. Exp. Bot. 2018, 69, 997–1009. [Google Scholar] [CrossRef]
- Naoumkina, M.; Thyssen, G.N.; Fang, D.D.; Li, P.; Florane, C.B. Elucidation of sequence polymorphism in fuzzless-seed cotton lines. Mol. Genet. Genom. MGG 2021, 296, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Chen, J.; Fang, L.; Zhang, Z.; Ma, W.; Niu, Y.; Ju, L.; Deng, J.; Zhao, T.; Lian, J.; et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 2019, 51, 739–748. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. Feature counts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Downs, G.S.; Bi, Y.M.; Colasanti, J.; Wu, W.; Chen, X.; Zhu, T.; Rothstein, S.J.; Lukens, L.N. A developmental transcriptional network for maize defines coexpression modules. Plant Physiol. 2013, 161, 1830–1843. [Google Scholar] [CrossRef] [Green Version]
- Deng, T.; Yao, H.; Wang, J. GhLTPG1, a cotton GPI-anchored lipid transfer protein, regulates the transport of phosphatidylinositol monophosphates and cotton fiber elongation. Sci. Rep. 2016, 6, 26829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, R.; Yuan, H.; An, J. A Gossypium hirsutum GDSL lipase/hydrolase gene (GhGLIP) appears to be involved in promoting seed growth in Arabidopsis. PLoS ONE 2018, 13, e0195556. [Google Scholar] [CrossRef] [Green Version]
- Molina, I.; Kosma, D. Role of HXXXD-motif/BAHD acyltransferases in the biosynthesis of extracellular lipids. Plant Cell Rep. 2014, 34, 587–601. [Google Scholar] [CrossRef]
- Hu, H.; He, X.; Tu, L. GhJAZ2 negatively regulates cotton fiber initiation by interacting with the R2R3-MYB transcription factor GhMYB25-like. Plant J. 2016, 88, 921–935. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Lou, P.; Han, Y. GrTCP11, a Cotton TCP Transcription Factor, Inhibits Root Hair Elongation by Down-Regulating Jasmonic Acid Pathway in Arabidopsis thaliana. Front. Plant Sci. 2021, 12, 769675. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.-Q.; Gong, S.-Y.; Xu, W.-L. A Fasciclin-Like Arabinogalactan Protein, GhFLA1, Is Involved in Fiber Initiation and Elongation of Cotton. Plant Physiol. 2013, 161, 1278–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Tu, L.; Li, Y. Genes Encoding Fasciclin-Like Arabinogalactan Proteins are Specifically Expressed During Cotton Fiber Development. Plant Mol. Biol. Rep. 2008, 26, 98–113. [Google Scholar] [CrossRef]
- Broun, P.; Poindexter, P.; Osborne, E.; Jiang, C.Z.; Riechmann, J.L. WIN1, a transcriptional activator of epidermal wax accumulation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2004, 101, 4706–4711. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.X.; Adato, A.; Alkan, N.; He, Y.; Lashbrooke, J.; Matas, A.J.; Meir, S.; Malitsky, S.; Isaacson, T.; Prusky, D.; et al. The tomato SlSHINE3 transcription factor regulates fruit cuticle formation and epidermal patterning. New Phytol. 2013, 197, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, A.; Dixit, S.; Jetter, R.; Thoenes, E.; Van, A.G.; Pereira, A. The SHINE clade of AP2 domain transcription factors activates wax biosynthesis, alters cuticle properties, and confers drought tolerance when overexpressed in Arabidopsis. Plant Cell 2004, 16, 2463–2480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, X.; Yan, S.; Tan, R.; Zhang, Z.; Wang, Z.; Chen, J. Characterization and expression of a GDSL-like lipase gene from Brassica napus in Nicotiana benthamiana. Protein J. 2014, 33, 18–23. [Google Scholar] [CrossRef]
- Agee, A.E.; Surpin, M.; Sohn, E.J.; Girke, T.; Rosado, A.; Kram, B.W.; Carter, C.; Wentzell, A.M.; Kliebenstein, D.; Jin, H.C.; et al. Modified vacuole phenotype1 is an Arabidopsis myrosinase-associated protein involved in endomembrane protein trafficking. Plant Physiol. 2010, 152, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Updegraff, E.P.; Zhao, F.; Preuss, D. The extracellular lipase EXL4 is required for efficient hydration of Arabidopsis pollen. Sex Plant Reprod. 2009, 22, 197–204. [Google Scholar] [CrossRef]
- Kim, K.J.; Lim, J.H.; Kim, M.J.; Kim, T.; Chung, H.M.; Paek, K.H. GDSL-lipase1 (CaGL1) contributes to wound stress resistance by modulation of CaPR-4 expression in hot pepper. Biochem. Biophys. Res. Commun. 2008, 374, 693–698. [Google Scholar] [CrossRef]
- Zhang, Z.; Ober, J.A.; Kliebenstein, D.J. The gene controlling the quantitative trait locus EPITHIOSPECIFIER MODIFIER1 alters glucosinolate hydrolysis and insect resistance in Arabidopsis. Plant Cell 2006, 18, 1524–1536. [Google Scholar] [CrossRef] [Green Version]
- Oh, I.S.; Park, A.R.; Bae, M.S.; Kwon, S.J.; Kim, Y.S.; Lee, J.E.; Kang, N.Y.; Lee, S.; Cheong, H.; Park, O.K. Secretome Analysis Reveals an Arabidopsis Lipase Involved in Defense against Alternaria brassicicola. Plant Cell 2005, 17, 2832–2847. [Google Scholar] [CrossRef] [Green Version]
- Ling, H.; Zhao, J.; Zuo, K.; Qiu, C.; Yao, H.; Qin, J.; Sun, X.; Tang, K. Isolation expression analysis of a GDSL -like lipase gene from Brassica napus, L. J. Biochem. Mol. Biol. 2006, 39, 297–303. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.P.; Huang, L.M.; Chen, L.O.; Chan, M.T.; Shaw, J.F. Genome-wide analysis of GDSL-type esterases/lipases in Arabidopsis. Plant Mol. Biol. 2017, 95, 181–197. [Google Scholar] [CrossRef]
- Dong, X.; Yi, H.; Han, C.T.; Nou, I.S.; Hur, Y. GDSL esterase/lipase genes in Brassica rapa L.: Genome-wide identification and expression analysis. Mol. Genet. Genom. 2016, 291, 531–542. [Google Scholar] [CrossRef]
- Chepyshko, H.; Lai, C.P.; Huang, L.M.; Liu, J.H.; Shaw, J.F. Multifunctionality and diversity of GDSL esterase/ lipase gene family in rice (Oryza sativa L. japonica) genome: New insights from bioinformatics analysis. BMC Genom. 2012, 13, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, S.J.; Jin, H.C.; Lee, S.; Nam, M.H.; Chung, J.H.; Kwon, S.I.; Ryu, C.-M.; Park, O.K. GDSL lipase-like 1 regulates systemic resistance associated with ethylene signaling in Arabidopsis. Plant J. 2009, 58, 235–245. [Google Scholar] [CrossRef]
- Lee, D.S.; Kim, B.K.; Kwon, S.J.; Jin, H.C.; Park, O.K. Arabidopsis GDSL lipase 2 plays a role in pathogen defense via negative regulation of auxin signaling. Biochem. Biophys. Res. Commun. 2009, 379, 1038–1042. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Jin, P.; Yoon, J.; Yang, J.-I.; Jeong, H.J.; Ranathunge, K.; Schreiber, L.; Franke, R.; Lee, I.-J.; An, G. Mutation in Wilted Dwarf and Lethal 1 (WDL1) causes abnormal cuticle formation and rapid water loss in rice. Plant Mol. Biol. 2010, 74, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.K.; Choi, H.W.; Hwang, I.S.; Kim, D.S.; Kim, N.H.; Choi, D.S.; Kim, Y.J.; Hwang, B.K. Function of a novel GDSL-type pepper lipase gene, CaGLIP1, in disease susceptibility and abiotic stress tolerance. Planta 2008, 227, 539–558. [Google Scholar] [CrossRef]
- Girard, A.-L.; Mounet, F.; Lemaire-Chamley, M.; Gaillard, C.; Elmorjani, K.; Vivancos, J.; Runavot, J.-L.; Quemener, B.; Petit, J.; Germain, V.; et al. Tomato GDSL1 is required for cutin deposition in the fruit cuticle. Plant Cell 2012, 24, 3119–3134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.P.; Tan, H.; Si, Y.; Creech, R.G.; Jenkins, J.N. Differential expression of a lipid transfer protein gene in cotton fiber. Bioch. Bioph. Acta 1995, 1257, 81–84. [Google Scholar] [CrossRef]
- Han, H.C.; Huang, Y.Q.; Wang, J.; Zuo, K.J. Cloning expression pattern analysis of Gossypium hirsutum lipid transfer protein gene family. J. Agric. Sci. Technol. 2013, 15, 84–90. [Google Scholar]
- Guan, X.Y.; Li, Q.J.; Shan, C.M.; Wang, S.; Mao, Y.B.; Wang, L.J.; Chen, X.Y. The HD-Zip IV gene GaHOX1 from cotton is a functional homologue of the Arabidopsis GLABRA2. Physiol. Plant 2008, 134, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Pu, L.; Li, Q.; Fan, X.P.; Yang, W.C.; Xue, Y.B. The R2R3 MYB transcription factor GhMYB109 is required for cotton fiber development. Genetics 2008, 180, 811–820. [Google Scholar] [CrossRef]
- Machado, A.; Wu, Y.R.; Yang, Y.M.; Llewellyn, D.J.; Dennis, E.S. The MYB transcription factor GhMYB25 regulates early fibre and trichome development. Plant J. 2009, 59, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Walford, S.A.; Wu, Y.R.; Llewellyn, D.J.; Dennis, E.S. GhMYB25-like: A key factor in early cotton fibre development. Plant J. 2011, 65, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Q.; Liu, X.; Tang, K.X.; Zuo, K.J. Functional analysis of the seed coat-specific gene GbMYB2 form cotton. Plant. Bioch. 2013, 73, 16–22. [Google Scholar] [CrossRef]
- Hsu, C.-Y.; Jenkins, J.N.; Saha, S.; Ma, D.-P. Transcriptional regulation of the lipid transfer protein gene LTP3 in cotton fiber by a novel MYB protein. Plant Sci. 2005, 168, 167–181. [Google Scholar] [CrossRef]


















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiao, Y.; Long, Y.; Xu, K.; Zhao, F.; Zhao, J.; Li, S.; Geng, S.; Gao, W.; Sun, P.; Deng, X.; et al. Weighted Gene Co-Expression Network Analysis Reveals Hub Genes for Fuzz Development in Gossypium hirsutum. Genes 2023, 14, 208. https://doi.org/10.3390/genes14010208
Jiao Y, Long Y, Xu K, Zhao F, Zhao J, Li S, Geng S, Gao W, Sun P, Deng X, et al. Weighted Gene Co-Expression Network Analysis Reveals Hub Genes for Fuzz Development in Gossypium hirsutum. Genes. 2023; 14(1):208. https://doi.org/10.3390/genes14010208
Chicago/Turabian StyleJiao, Yang, Yilei Long, Kaixiang Xu, Fuxiang Zhao, Jieyin Zhao, Shengmei Li, Shiwei Geng, Wenju Gao, Peng Sun, Xiaojuan Deng, and et al. 2023. "Weighted Gene Co-Expression Network Analysis Reveals Hub Genes for Fuzz Development in Gossypium hirsutum" Genes 14, no. 1: 208. https://doi.org/10.3390/genes14010208