
Altered Left Ventricular Rat Gene Expression Induced by the Myosin Activator Omecamtiv Mecarbil
 , , ,
, , ,
Abstract
:1. Introduction
2. Methods
2.1. Animal Model and Experimental Design
2.2. Echocardiography and Cardiac Measurements
2.3. Real-Time Quantitative Polymerase Chain Reaction (RTq-PCR)
2.4. Statistical Analysis
3. Results
3.1. Effect of OM on Cardiac Function in Rats
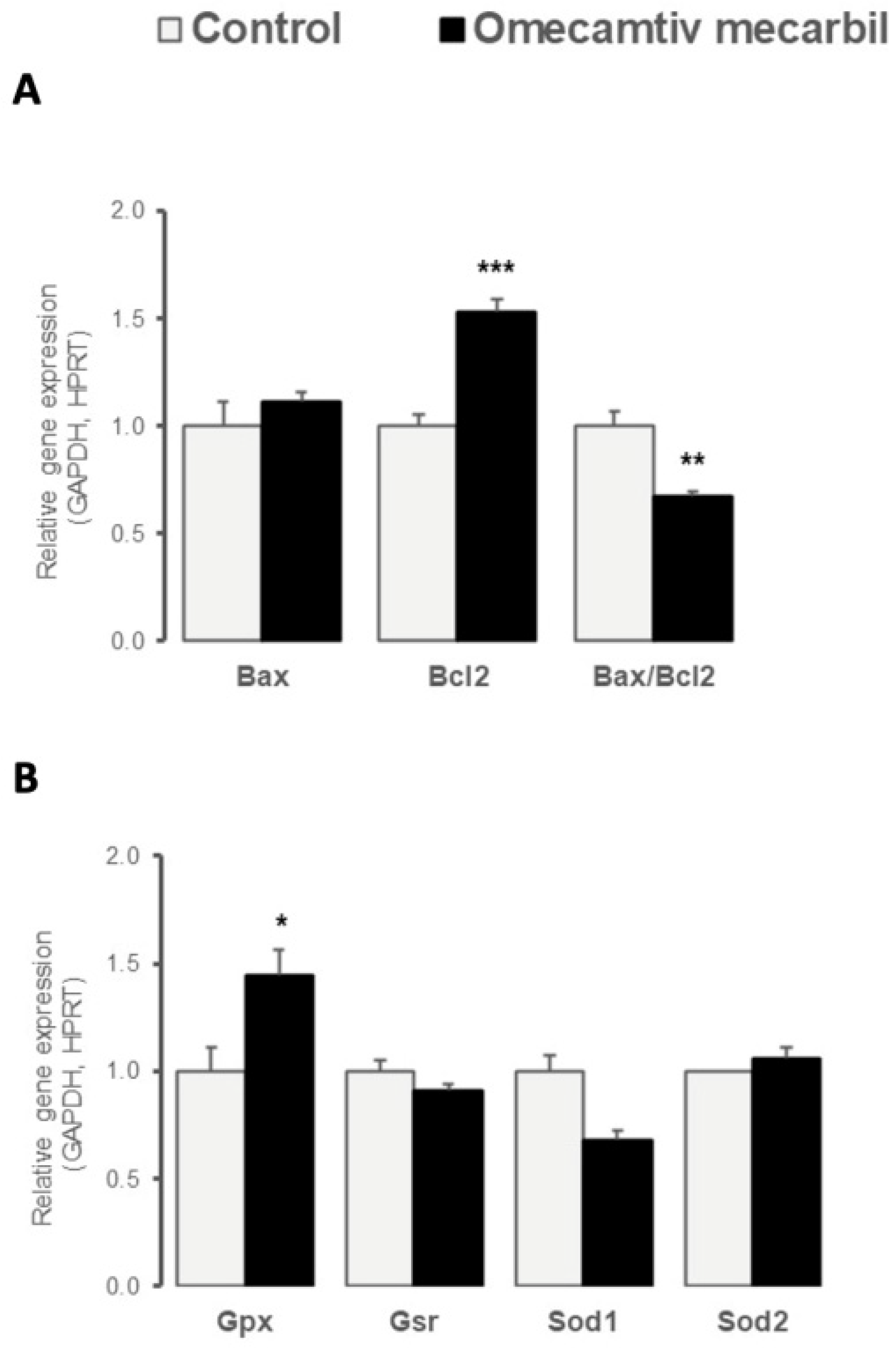
3.2. OM Altered Myocardial LV Expression of Genes Regulating Apoptosis and Oxidative Stress
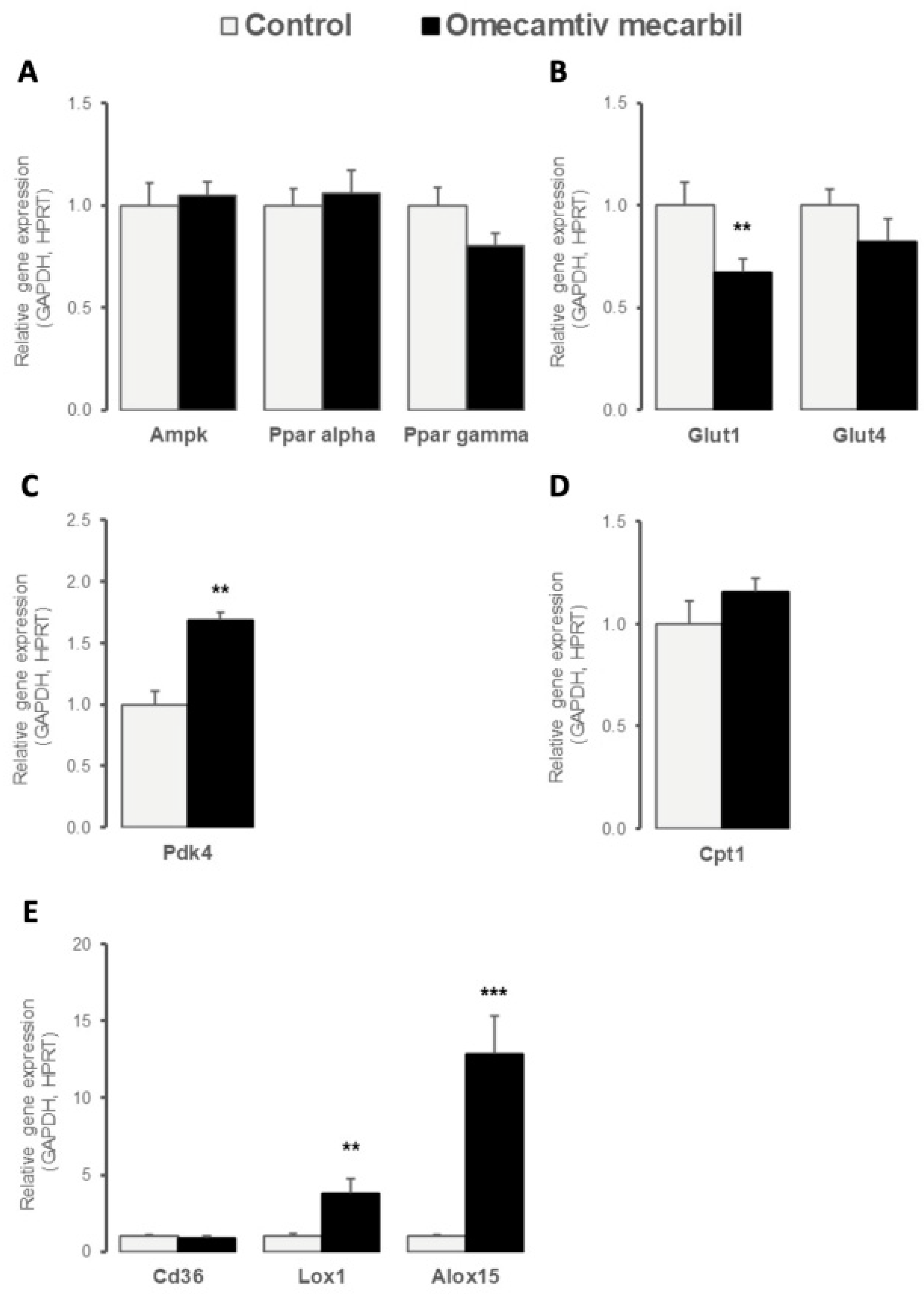
3.3. OM Impacted LV Expression Profile of Key Determinants of Cardiac Energy Substrate Use
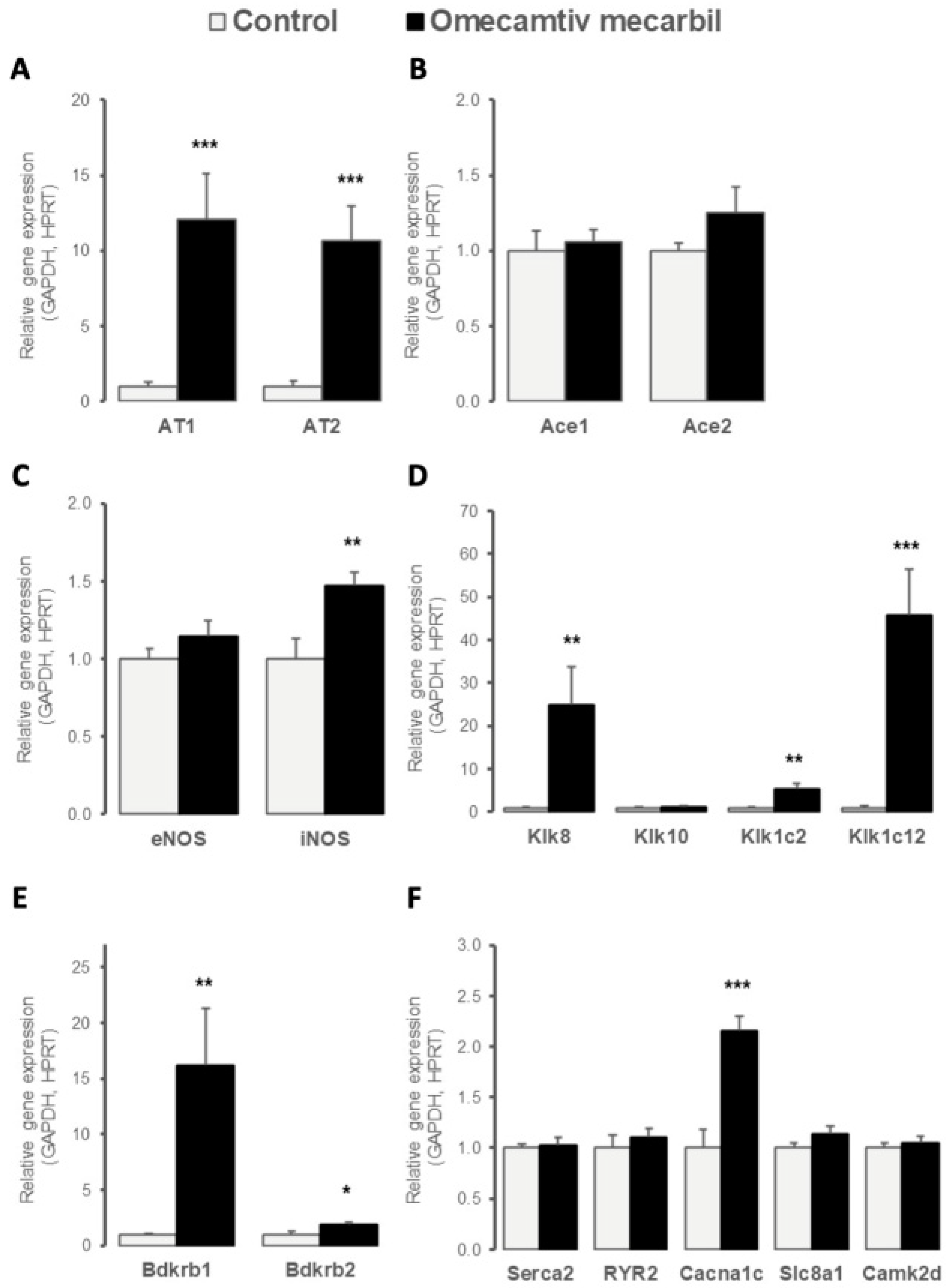
3.4. OM Altered LV Expression of Genes Implicated in Cardiac Contractility
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; de Ferranti, S.; Després, J.P.; Fullerton, H.J.; Howard, V.J.; et al. Heart disease and stroke statistics—2015 update: A report from the American Heart Association. Circulation 2015, 131, e29–e322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, K.F., Jr.; Fonarow, G.C.; Emerman, C.L.; LeJemtel, T.H.; Costanzo, M.R.; Abraham, W.T.; Berkowitz, R.L.; Galvao, M.; Horton, D.P.; ADHERE Scientific Advisory Committee and Investigators. Characteristics and outcomes of patients hospitalized for heart failure in the United States: Rationale, design, and preliminary observations from the first 100,000 cases in the Acute Decompensated Heart Failure National Registry (ADHERE). Am. Heart J. 2005, 149, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Follath, F.; Cleland, J.G.; Just, H.; Papp, J.G.; Scholz, H.; Peuhkurinen, K.; Harjola, V.P.; Mitrovic, V.; Abdalla, M.; Sandell, E.P.; et al. Efficacy and safety of intravenous levosimendan compared with dobutamine in severe low-output heart failure (the LIDO study): A randomised double-blind trial. Steering Committee and Investigators of the Levosimendan Infusion versus Dobutamine (LIDO) Study. Lancet 2002, 360, 196–202. [Google Scholar] [CrossRef]
- Song, L.S.; Wang, S.Q.; Xiao, R.P.; Spurgeon, H.; Lakatta, E.G.; Cheng, H. beta-Adrenergic stimulation synchronizes intracellular Ca(2+) release during excitation-contraction coupling in cardiac myocytes. Circ Res. 2001, 88, 794–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bristow, M.R. Treatment of chronic heart failure with beta-adrenergic receptor antagonists: A convergence of receptor pharmacology and clinical cardiology. Circ Res 2011, 109, 1176–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stapel, B.; Kohlhaas, M.; Ricke-Hoch, M.; Haghikia, A.; Erschow, S.; Knuuti, J.; Silvola, J.M.U.; Roivainen, A.; Saraste, A.; Nickel, A.G.; et al. Low STAT3 expression sensitizes to toxic effects of β-adrenergic receptor stimulation in peripartum cardiomyopathy. Eur. Heart J. 2017, 38, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Malik, F.I.; Hartman, J.J.; Elias, K.A.; Morgan, B.P.; Rodriguez, H.; Brejc, K.; Anderson, R.L.; Sueoka, S.H.; Lee, K.H.; Finer, J.T.; et al. Cardiac Myosin Activation: A Potential Therapeutic Approach for Systolic Heart Failure. Science 2011, 331, 1439–1443. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.-T.; Malik, F.I.; Zhao, X.; Depre, C.; Dhar, S.K.; Abarzúa, P.; Morgans, D.J.; Vatner, S.F. Improvement of Cardiac Function by a Cardiac Myosin Activator in Conscious Dogs With Systolic Heart Failure. Circ. Heart Fail. 2010, 3, 522–527. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; White, H.D.; Belknap, B.; Winkelmann, D.A.; Forgacs, E. Omecamtiv Mecarbil Modulates the Kinetic and Motile Properties of Porcine β-Cardiac Myosin. Biochemistry 2015, 54, 1963–1975. [Google Scholar] [CrossRef]
- Bakkehaug, J.P.; Kildal, A.B.; Engstad, E.T.; Boardman, N.; Næsheim, T.; Rønning, L.; Aasum, E.; Larsen, T.S.; Myrmel, T.; How, O.-J. Myosin Activator Omecamtiv Mecarbil Increases Myocardial Oxygen Consumption and Impairs Cardiac Efficiency Mediated by Resting Myosin ATPase Activity. Circ. Heart Fail. 2015, 8, 766–775. [Google Scholar] [CrossRef]
- Nagy, L.; Kovács, A.; Bódi, B.; Pásztor, E.T.; Fülöp, G.; Tóth, A.; Édes, I.; Papp, Z. The novel cardiac myosin activator omecamtiv mecarbil increases the calcium sensitivity of force production in isolated cardiomyocytes and skeletal muscle fibres of the rat. Br. J. Pharmacol. 2015, 172, 4506–4518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utter, M.S.; Ryba, D.; Li, B.H.; Wolska, B.M.; Solaro, R.J. Omecamtiv Mecarbil, a Cardiac Myosin Activator, Increases Ca2+ Sensitivity in Myofilaments With a Dilated Cardiomyopathy Mutant Tropomyosin E54K. J. Cardiovasc. Pharmacol. 2015, 66, 347–353. [Google Scholar] [CrossRef]
- Anderson, R.L.; Sueoka, S.H.; Rodriguez, H.M.; Lee, K.H.; Cox, D.R.; Kawas, R.; Morgan, B.P.; Sakowicz, R.; Morgans, D.J.; Malik, F.; et al. In vitro and in vivo efficacy of the cardiac myosin activator CK-1827452. Mol Bio Cell 2005, 16. Available online: https://cytokinetics.com/wp-content/uploads/2015/10/ASCB_1728.pdf (accessed on 19 December 2022).
- Nagueh, S.F.; Smiseth, O.A.; Appleton, C.P.; Byrd, B.F., 3rd; Dokainish, H.; Edvardsen, T.; Flachskampf, F.A.; Gillebert, T.C.; Klein, A.L.; Lancellotti, P.; et al. Recommendations for the Evaluation of Left Ventricular Diastolic Function by Echocardiography: An Update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. 2016, 29, 277–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaffla, M.W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- El-Oumeiri, B.; Mc Entee, K.; Annoni, F.; Herpain, A.; Eynden, F.V.; Jespers, P.; Van Nooten, G.; Van De Borne, P. Effects of the cardiac myosin activator Omecamtiv-mecarbil on severe chronic aortic regurgitation in Wistar rats. BMC Cardiovasc. Disord. 2018, 18, 99. [Google Scholar] [CrossRef] [PubMed]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Machado, A.R.T.; Aissa, A.F.; Ribeiro, D.L.; Hernandes, L.C.; Machado, C.S.; Bianchi, M.L.P.; Sampaio, S.V.; Antunes, L.M.G. The toxin BjussuLAAO-II induces oxidative stress and DNA damage, upregulates the inflammatory cytokine genes TNF and IL6, and downregulates the apoptotic-related genes BAX, BCL2 and RELA in human Caco-2 cells. Int. J. Biol. Macromol. 2018, 109, 212–219. [Google Scholar] [CrossRef]
- Jarskog, L.F.; Selinger, E.S.; Lieberman, J.A.; Gilmore, J.H. Apoptotic proteins in the temporal cortex in schizophrenia: High Bax/Bcl-2 ratio without caspase-3 activation. Am. J. Psychiatry 2004, 161, 109–115. [Google Scholar] [CrossRef] [Green Version]
- Dostanic, S.; Servant, N.; Wang, C.; E Chalifour, L. Chronic β-adrenoreceptor stimulation in vivo decreased Bcl-2 and increased Bax expression but did not activate apoptotic pathways in mouse heart. Can. J. Physiol. Pharmacol. 2004, 82, 167–174. [Google Scholar] [CrossRef]
- Manoharan, S.; Kolanjiappan, K.; Suresh, K.; Panjamurthy, K. Lipid peroxidation & antioxidants status in patients with oral squamous cell carcinoma. Indian J. Med. Res. 2005, 122, 529–534. [Google Scholar]
- Gozeler, M.S.; Akdemir, F.N.E.; Yildirim, S.; Sahin, A.; Eser, G.; Askin, S. Levosimendan ameliorates cisplatin-induced ototoxicity: Rat model. Int. J. Pediatr. Otorhinolaryngol. 2019, 122, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Rhoden, A.; Schulze, T.; Pietsch, N.; Christ, T.; Hansen, A.; Eschenhagen, T. Comprehensive analyses of the inotropic compound omecamtiv mecarbil in rat and human cardiac preparations. Am. J. Physiol. Circ. Physiol. 2022, 322, H373–H385. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Dandapat, A.; Chen, J.; Fujita, Y.; Inoue, N.; Kawase, Y.; Jishage, K.; Suzuki, H.; Sawamura, T.; Mehta, J.L. LOX-1 deletion alters signals of myocardial remodeling immediately after ischemia-reperfusion. Cardiovasc. Res. 2007, 76, 292–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egert, S.; Nguyen, N.; Schwaiger, M. Contribution of α-Adrenergic and β-Adrenergic Stimulation to Ischemia-Induced Glucose Transporter (GLUT) 4 and GLUT1 Translocation in the Isolated Perfused Rat Heart. Circ. Res. 1999, 84, 1407–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, J.L.; Stanley, W.C.; Lopaschuk, G.D.; Wisneski, J.A.; Pizzurro, R.D.; Hamilton, C.D.; McCormack, J.G. Impaired pyruvate oxidation but normal glucose uptake in diabetic pig heart during dobutamine-induced work. Am. J. Physiol. Circ. Physiol. 1996, 271, H2320–H2329. [Google Scholar] [CrossRef]
- Sugden, M.C.; Holness, M.J. Interactive regulation of the pyruvate dehydrogenase complex and the carnitine palmitoyltransferase system. FASEB J. 1994, 8, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Sato, J.; Zhao, Y.; Jaskiewicz, J.; Popov, M.K.; Harris, A.R. Starvation and diabetes increase the amount of pyruvate dehydrogenase kinase isoenzyme 4 in rat heart. Biochem. J. 1998, 329, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, I.K.N.; Tusubira, D.; Ashrafi, H.; Dyrstad, S.E.; Hansen, L.; Liu, X.Z.; Nilsson, L.I.H.; Løvsletten, N.G.; Berge, K.; Wergedahl, H.; et al. Upregulated PDK4 expression is a sensitive marker of increased fatty acid oxidation. Mitochondrion 2019, 49, 97–110. [Google Scholar] [CrossRef]
- Sawamura, T.; Kume, N.; Aoyama, T.; Moriwaki, H.; Hoshikawa, H.; Aiba, Y.; Tanaka, T.; Miwa, S.; Katsura, Y.; Kita, T.; et al. An endothelial receptor for oxidized low-density lipoprotein. Nature 1997, 386, 73–77. [Google Scholar] [CrossRef]
- Li, D.Y.; Zhang, Y.C.; Philips, M.I.; Sawamura, T.; Mehta, J.L. Upregulation of Endothelial Receptor for Oxidized Low-Density Lipoprotein (LOX-1) in Cultured Human Coronary Artery Endothelial Cells by Angiotensin II Type 1 Receptor Activation. Circ. Res. 1999, 84, 1043–1049. [Google Scholar] [CrossRef] [Green Version]
- Murase, T.; Kume, N.; Korenaga, R.; Ando, J.; Sawamura, T.; Masaki, T.; Kita, T. Fluid Shear Stress Transcriptionally Induces Lectin-like Oxidized LDL Receptor-1 in Vascular Endothelial Cells. Circ. Res. 1998, 83, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Williams, V.; Liu, L.; Chen, H.; Sawamura, T.; Antakli, T.; Mehta, J.L. Mehta LOX-1 inhibition in myocardial ischemia-reperfusion injury: Modulation of MMP-1 and inflammation. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1795–H1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, H.; O’Donnell, V.B. Inflammation and immune regulation by 12/15-lipoxygenases. Prog. Lipid Res. 2006, 45, 334–356. [Google Scholar] [CrossRef] [PubMed]
- Kayama, Y.; Minamino, T.; Toko, H.; Sakamoto, M.; Shimizu, I.; Takahashi, H.; Okada, S.; Tateno, K.; Moriya, J.; Yokoyama, M.; et al. Cardiac 12/15 lipoxygenase–induced inflammation is involved in heart failure. J. Exp. Med. 2009, 206, 1565–1574. [Google Scholar] [CrossRef] [Green Version]
- Takaya, T.; Wada, H.; Morimoto, T.; Sunagawa, Y.; Kawamura, T.; Takanabe-Mori, R.; Shimatsu, A.; Fujita, Y.; Sato, Y.; Fujita, M.; et al. Left Ventricular Expression of Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1 in Failing Rat Hearts. Circ. J. 2010, 74, 723–729. [Google Scholar] [CrossRef] [Green Version]
- Zendaoui, A.; Lachance, D.; Roussel, E.; Couet, J.; Arsenault, M. Effects of spironolactone treatment on an experimental model of chronic aortic valve regurgitation. J. Heart Valve Dis. 2012, 21, 478–486. [Google Scholar]
- Nánási, P., Jr.; Gaburjakova, M.; Gaburjakova, J.; Almássy, J. Omecamtiv mecarbil activates ryanodine receptors from canine cardiac but not skeletal muscle. Eur. J. Pharmacol. 2017, 809, 73–79. [Google Scholar] [CrossRef]
- Fülöp, G.; Oláh, A.; Csipo, T.; Kovács, Á.; Pórszász, R.; Veress, R.; Horváth, B.; Nagy, L.; Bódi, B.; Fagyas, M.; et al. Omecamtiv mecarbil evokes diastolic dysfunction and leads to periodic electromechanical alternans. Basic Res. Cardiol. 2021, 116, 24. [Google Scholar] [CrossRef]
- Cassis, P.; Conti, S.; Remuzzi, G.; Benigni, A. Angiotensin receptors as determinants of life span. Pflügers Arch.-Eur. J. Physiol. 2010, 459, 325–332. [Google Scholar] [CrossRef]
- Namsolleck, P.; Recarti, C.; Foulquier, S.; Steckelings, U.M.; Unger, T. AT(2) receptor and tissue injury: Therapeutic implications. Curr. Hypertens. Rep. 2014, 16, 416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobales, N.; Nuñez, R.E.; Javadov, S. Mitochondrial angiotensin receptors and cardioprotective pathways. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1426–H1438. [Google Scholar] [CrossRef]
- Cho, H.J.; Xie, Q.W.; Calaycay, J.; Mumford, R.A.; Swiderek, K.M.; Lee, T.D.; Nathan, C. Calmodulin is a subunit of nitric oxide synthase from macrophages. J. Exp. Med. 1992, 176, 599–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziolo, M.T.; Harshbarger, C.H.; Roycroft, K.E.; Smith, J.M.; Romano, F.D.; Sondgeroth, K.L.; Wahler, G.M. Myocytes Isolated from Rejecting Transplanted Rat Hearts Exhibit a Nitric Oxide-mediated Reduction in the Calcium Current. J. Mol. Cell. Cardiol. 2001, 33, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Wildhirt, S.M.; Weismueller, S.; Schulze, C.; Conrad, N.; Kornberg, A.; Reichart, B. Inducible nitric oxide synthase activation after ischemia/reperfusion contributes to myocardial dysfunction and extent of infarct size in rabbits: Evidence for a late phase of nitric oxide-mediated reperfusion injury. Cardiovasc. Res. 1999, 43, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Ziolo, M.T.; Maier, L.S.; Piacentino, V., 3rd; Bossuyt, J.; Houser, S.R.; Bers, D.M. Myocyte nitric oxide synthase 2 contributes to blunted beta-adrenergic response in failing human hearts by decreasing Ca2+ transients. Circulation 2004, 109, 1886–1891. [Google Scholar] [CrossRef] [Green Version]
- Heymes, C.; Vanderheyden, M.; Bronzwaer, J.G.; Shah, A.M.; Paulus, W.J. Endomyocardial nitric oxide synthase and left ventricular preload reserve in dilated cardiomyopathy. Circulation 1999, 99, 3009–3016. [Google Scholar] [CrossRef] [Green Version]
- Paulus, W.J. Beneficial effects of nitric oxide on cardiac diastolic function: ‘The flip side of the coin’. Heart Fail. Rev. 2000, 5, 337–344. [Google Scholar] [CrossRef]
- Cao, B.; Yu, Q.; Zhao, W.; Tang, Z.; Cong, B.; Du, J.; Lu, J.; Zhu, X.; Ni, X. Kallikrein-related peptidase 8 is expressed in myocardium and induces cardiac hypertrophy. Sci. Rep. 2016, 7, 20024. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Du, J.; Wang, Y.; Ma, S.; Hu, T.; Shang, J.; Yu, Q.; Zhu, X.; Zhang, G.; Cong, B. Tissue kallikrein-related peptidase8 protects rat heart against acute ischemia reperfusion injury. Int. J. Biol. Macromol. 2019, 140, 1126–1133. [Google Scholar] [CrossRef]
- Rougeot, C.; Rosinski-Chupin, I.; Mathison, R.; Rougeon, F. Rodent submandibular gland peptide hormones and other biologically active peptides. Peptides 2000, 21, 443–455. [Google Scholar] [CrossRef]
- Borges, J.C.; Silva, J.; Gomes, M.A.; Lomez, E.S.L.; Leite, K.M.; Araujo, R.C.; Bader, M.; Pesquero, J.B.; Pesquero, J.L. Tonin in rat heart with experimental hypertrophy. Am. J. Physiol. Circ. Physiol. 2003, 284, H2263–H2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damasceno, D.D.; Lima, M.P.; Motta, D.F.; Ferreira, A.J.; Quintão-Junior, J.F.; Drummond, L.R.; Natali, A.J.; Almeida, A.P.; Pesquero, J.L. Cardiovascular and electrocardiographic parameters after tonin administration in Wistar rats. Regul. Pept. 2013, 181, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Shen, B.; Gao, L.; Xia, C.-F.; Bledsoe, G.; Chao, L. Tissue kallikrein in cardiovascular, cerebrovascular and renal diseases and skin wound healing. Biol. Chem. 2010, 391, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Hamid, S.; Rhaleb, I.; Kassem, K.; Rhaleb, N.-E. Role of Kinins in Hypertension and Heart Failure. Pharmaceuticals 2020, 13, 347. [Google Scholar] [CrossRef] [PubMed]
- Abadir, P.M.; Periasamy, A.; Carey, R.M.; Siragy, H.M. Angiotensin II type 2 receptor-bradykinin B2 receptor functional heterodimerization. Hypertension 2006, 48, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Levy, R.F.; Serra, A.J.; Antonio, E.L.; Dos Santos, L.; Bocalini, D.S.; Pesquero, J.B.; Bader, M.; Merino, V.F.; De Oliveira, H.A.; Veiga, E.C.D.A.; et al. Cardiac Morphofunctional Characteristics of Transgenic Rats with Overexpression of the Bradykinin B1 Receptor in the Endothelium. Physiol. Res. 2017, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Duka, A.; Kintsurashvili, E.; Duka, I.; Ona, D.; Hopkins, T.A.; Bader, M.; Gavras, I.; Gavras, H. Angiotensin-converting enzyme inhibition after experimental myocardial infarct: Role of the kinin B1 and B2 receptors. Hypertension 2008, 51, 1352–1357. [Google Scholar] [CrossRef] [Green Version]
- Zamponi, G.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [Green Version]
- Westhoff, M.; Dixon, R.E. Mechanisms and Regulation of Cardiac CaV1.2 Trafficking. Int. J. Mol. Sci. 2021, 22, 5927. [Google Scholar] [CrossRef]
- Szentandrassy, N.; Horvath, B.; Vaczi, K.; Kistamas, K.; Masuda, L.; Magyar, J.; Banyasz, T.; Papp, Z.; Nanasi, P.P. Dose-dependent electrophysiological effects of the myosin activator omecamtiv mecarbil in canine ventricular cardiomyocytes. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2016, 67, 483–489. [Google Scholar]
- Christel, C.J.; Cardona, N.; Mesirca, P.; Herrmann, S.; Hofmann, F.; Striessnig, J.; Ludwig, A.; Mangoni, M.E.; Lee, A. Distinct localization and modulation of Cav1.2 and Cav1.3 L-type Ca2+ channels in mouse sinoatrial node. J. Physiol. 2012, 590, 6327–6342. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Genes | Primer Sequences | |
|---|---|---|
| Glycerol-3-phosphate dehydrogenase (GAPDH) | Sense Antisense | 5′-AAGATGGTGAAGGTCGGTGT-3′ 5′-ATGAAGGGGTCGTTGATGG-3′ |
| Hypoxanthine guanine phosphoribosyl transferase (HPRT) | Sense Antisense | 5′-ACAGGCCAGACTTTGTTGGA-3′ 5′-ATCCACTTTCGCTGATGACAC-3′ |
| AMP-activated protein kinase (Ampk) | Sense Antisense | 5′-TTCGGGAAAGTGAAGGTGGG-3′ 5′-TCTCTGCGGATTTTCCCGAC-3′ |
| Angiotensin-converting enzyme 1 (ACE1) | Sense Antisense | 5′-AGTGGGTGCTGCTCTTCCTA-3′ 5′-GGAGGCTGTGATGGTTATGG-3′ |
| Angiotensin-converting enzyme 2 (ACE2) | Sense Antisense | 5′-GCCTTGGAAAATGTGGTAGG-3′ 5′-TTCAGCCAGACAAACAATGG-3′ |
| Angiotensin II receptor type 1a (Agtr1a or AT1) | Sense Antisense | 5′-ACATTCTGGGCTTCGTGTTC-3′ 5′-CATCATTTCTTGGCGTGTTC-3′ |
| Angiotensin II receptor type 2 (Agtr2 or AT2) | Sense Antisense | 5′-TGCTCTGACCTGGATGGGTA-3′ 5′-AGCTGTTTGGTGAATCCCAGG-3′ |
| Arachidonate 15-lipoxygenase (Alox15) | Sense Antisense | 5′-GCACTCTTCCGTCCATCTTG-3′ 5′-GCTTCTCCATTGTTGCTTCCT-3′ |
| ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 (Atp2a2 or Serca2) | Sense Antisense | 5′-GCAGGTCAAGAAGCTCAAGG-3′ 5′-TCTCTGCGGATTTTCCCGAC-3′ |
| Bcl2 associated X apoptosis regulator (Bax) | Sense Antisense | 5′-CGTGGTTGCCCTCTTCTACT-3′ 5′-TCACGGAGGAAGTCCAGTGT-3′ |
| B-cell lymphoma 2 (Bcl2) | Sense Antisense | 5′-TTTCTCCTGGCTGTCTCTGAA-3′ 5′-CATATTTGTTTGGGGCAGGT-3′ |
| Bradykinin receptor B1 (Bdkrb1) | Sense Antisense | 5′-AAGCTACGTGCCTGCTCATC-3′ 5′-CGGGGACGACTTTAACAGAG-3′ |
| Bradykinin receptor B2 (Bdkrb2) | Sense Antisense | 5′-GCTGTCGTGGAAGTGGCTAT-3′ 5′-AAGGTCCCGTTATGAGCAGA-3′ |
| Ca2+/calmodulin-dependent protein kinase II delta (Camk2d) | Sense Antisense | 5′-ATCCACAACCCTGATGGAAA-3′ 5′-GCTTTCGTGTTTCACGTCT-3′ |
| Ca2+ voltage-gated channel subunit alpha1 C (Cacna1c) | Sense Antisense | 5′-CCTATTTCCGTGACCTGTGG-3′ 5′-GGAGGGACTTGATGGTGTTG-3′ |
| Carnitine palmitoyltransferase 1 (Cpt1) | Sense Antisense | 5′-AAGAACACGAGCCAACAAGC-3′ 5′ACCATACCCAGTGCCATCAC-3′ |
| CD36 fatty acid transporter (Cd36) | Sense Antisense | 5′-TTTCTGCTTTCTCATCGCCG-3′ 5′-GGATGTGGAACCCATAACTGG-3′ |
| Glutathione peroxidase (Gpx) | Sense Antisense | 5′-CCGACCCCAAGTACATCATT-3′ 5′-AACACCGTCTGGACCTACCA-3′ |
| Glutathione-disulfide reductase (Gsr) | Sense Antisense | 5′-GCCGCCTGAACAACATCTAC-3′ 5′-CTTTTTCCCGTTGACTTCCA-3′ |
| Kallikrein-related peptidase 8 (Klk8) | Sense Antisense | 5′-CGGAGACAGATGGGTCCTAA-3′ 5′-ATCTCTTGCTCGGGCTCAT-3′ |
| Kallikrein-related peptidase 10 (Klk10) | Sense Antisense | 5′-GCAGGTCTCCCTCTTCCATA-3′ 5′-CAGTGGCTTATTTCTCCAGCA-3′ |
| Kallikrein 1-related peptidase C2 (Klk1c2) | Sense Antisense | 5′- CAGGAGAGATGGAAGGAGGA-3′ 5′-CGGTGTTTTGGGTTTAGCAC-3′ |
| Kallikrein 1-related peptidase C12 (Klk1c12) | Sense Antisense | 5′-CATCAAAGCCCACACACAGAT-3′ 5′-AAGCACACCATCACAGAGGAG-3′ |
| Nitric oxide synthase 2 (NOS2 or iNOS) | Sense Antisense | 5′-GTTTCCCCCAGATCCTCACT-3′ 5′-CTCTCCATTGCCCCAGTTT-3′ |
| Nitric oxide synthase 3 (NOS3 or eNOS) | Sense Antisense | 5′-GGTATTTGATGCTCGGGACT-3′ 5′-TGATGGCTGAACGAAGATTG-3′ |
| Oxidized low density lipoprotein receptor 1 (Olr1 or Lox1) | Sense Antisense | 5′-CATTCACCTCCCCATTTT-3′ 5′-GTAAAGAAACGCCCCTGGT-3′ |
| Peroxisome proliferator-activated receptor α (Ppar α) | Sense Antisense | 5′-TTAGAGGCGAGCCAAGACTG-3′ 5′-CAGAGCACCAATCTGTGATGA-3′ |
| Peroxisome proliferator-activated receptor γ (Ppar γ) | Sense Antisense | 5′– GCGCTAAATTCATCTTAACTC-3′ 5′-CTGTGTCAACCATGGTAATTT-3′ |
| Pyruvate dehydrogenase kinase 4 (Pdk4) | Sense Antisense | 5′-GAGCCTGATGGATTTAGTGGA-3′ 5′-CGAACTTTGACCAGCGTGT-3′ |
| Ryanodine receptor 2 (Ryr2) | Sense Antisense | 5′-GGAACTGACGGAGGAAAGTG-3′ 5′-GAGACCAGCATTTGGGTTGT-3′ |
| Solute carrier family 2 member 1 (Slc2a1 or Glut1) | Sense Antisense | 5′-TCTTCGAGAAGGCAGGTGTG-3′ 5′-TCCACGACGAACAGCGAC-3′ |
| Solute carrier family 2 member 4 (Slc2a4 or Glut4) | Sense Antisense | 5′-AGGCCGGGACACTATACCC-3′ 5′-TCCCCATCTTCAGAGCCGAT -5′ |
| Solute carrier family 8 member A1 (Slc8a1) | Sense Antisense | 5′-GAGATTGGAGAACCCCGTCT-3′ 5′-AGTGGCTGCTTGTCATCGTA-3′ |
| Superoxide dismutase 1 (Sod1) | Sense Antisense | 5′-GGTCCACGAGAAACAAGATGA-3′ 5′-CAATCACACCACAAGCCAAG-3′ |
| Superoxide dismutase 2 (Sod2) | Sense Antisense | 5′-AAGGAGCAAGGTCGCTTACA-3′ 5′-ACACATCAATCCCCAGCAGT-3′ |
| Parameters | Before OM Infusion | After OM Infusion |
|---|---|---|
| FS (%) | 38.8 ± 3.6 | 44.1 ± 4.4 * |
| EF (%) | 76.0 ± 4.4 | 80.6 ± 5.2 |
| LVESD (mm) | 5.5 ± 0.5 | 4.6 ± 0.9 |
| LVEDD (mm) | 9.0 ± 0.4 | 8.3 ± 0.9 |
| HR (beats/min) | 283 ± 57 | 303 ± 68 |
| SBP (mmHg) | 128 ± 9 | 128 ± 21 |
| DBP (mmHg) | 90 ± 10 | 88 ± 20 |
| LVET (ms) | 79 ± 6 | 89 ± 8 * |
| PEP (ms) | 21 ± 5 | 14 ± 7 * |
| PEP/LVET | 0.26 ± 0.06 | 0.15 ± 0.07 * |
| CO (mL/min) | 90 ± 21 | 106 ± 22 |
| SV (mL) | 0.32 ± 0.06 | 0.35 ± 0.04 |
| LA (mm) | 5.8 ± 0.8 | 5.7 ± 0.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Oumeiri, B.; Dewachter, L.; Van de Borne, P.; Hubesch, G.; Melot, C.; Jespers, P.; Stefanidis, C.; Mc Entee, K.; Vanden Eynden, F. Altered Left Ventricular Rat Gene Expression Induced by the Myosin Activator Omecamtiv Mecarbil. Genes 2023, 14, 122. https://doi.org/10.3390/genes14010122
El Oumeiri B, Dewachter L, Van de Borne P, Hubesch G, Melot C, Jespers P, Stefanidis C, Mc Entee K, Vanden Eynden F. Altered Left Ventricular Rat Gene Expression Induced by the Myosin Activator Omecamtiv Mecarbil. Genes. 2023; 14(1):122. https://doi.org/10.3390/genes14010122
Chicago/Turabian StyleEl Oumeiri, Bachar, Laurence Dewachter, Philippe Van de Borne, Géraldine Hubesch, Christian Melot, Pascale Jespers, Constantin Stefanidis, Kathleen Mc Entee, and Frédéric Vanden Eynden. 2023. "Altered Left Ventricular Rat Gene Expression Induced by the Myosin Activator Omecamtiv Mecarbil" Genes 14, no. 1: 122. https://doi.org/10.3390/genes14010122