

Frequency of Pathogenic Germline Mutations in Early and Late Onset Familial Breast Cancer Patients Using Multi-Gene Panel Sequencing: An Egyptian Study
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Methods
2.1. Patient Selection
2.2. DNA Extraction
2.3. NGS Assay
2.4. Bioinformatics Analysis
2.5. Statistical Analysis
3. Results
3.1. Patient Features
3.2. NGS Dataset Description
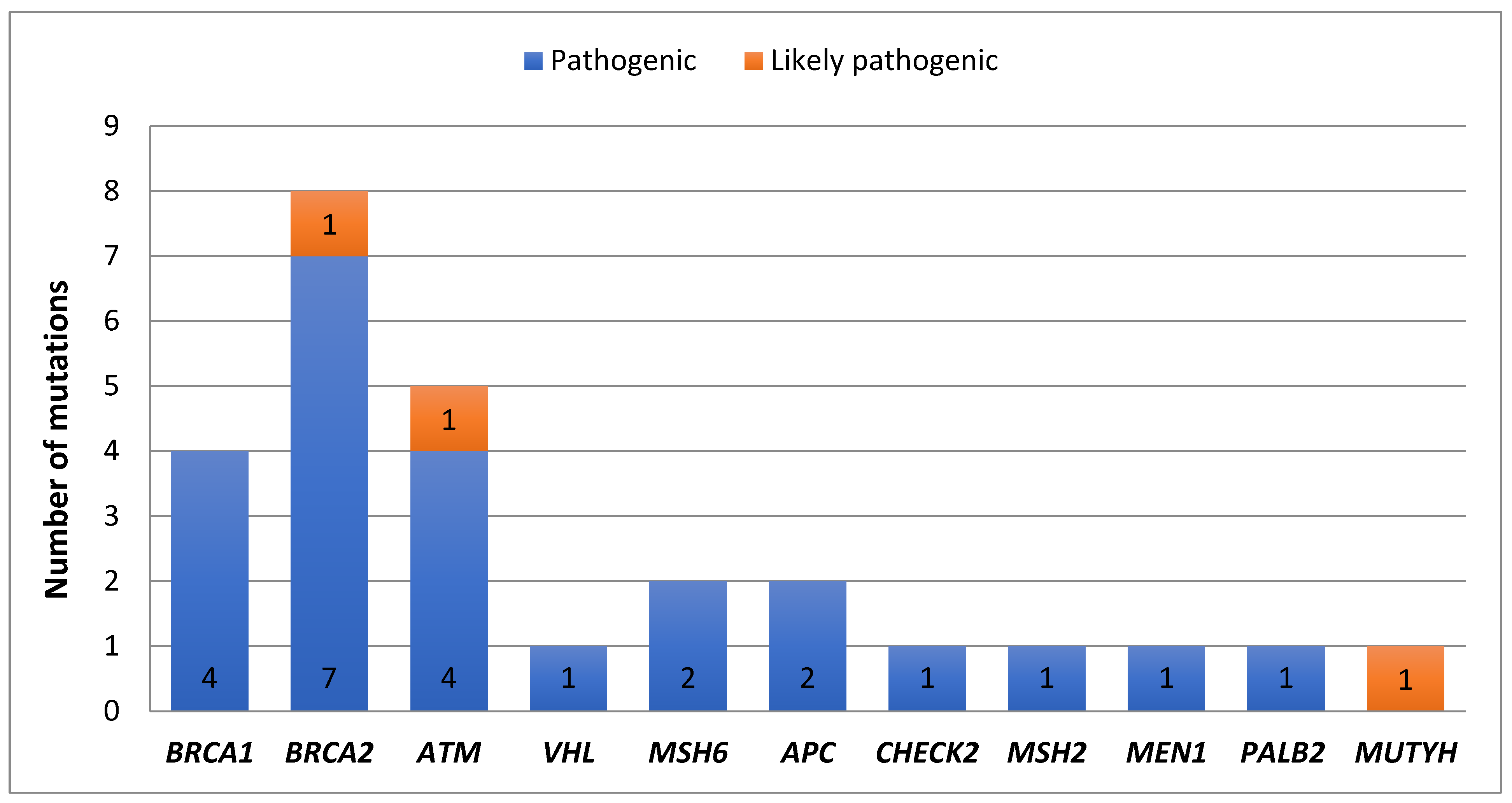
3.3. Frequency of Pathogenic Mutations Identified in This Cohort
3.4. Frequency of BRCA1/2 and Other DNA Repair Genes Identified in This Cohort
3.5. Frequency of Non-BRCA Genes in BRCA1/2 Negative Patients
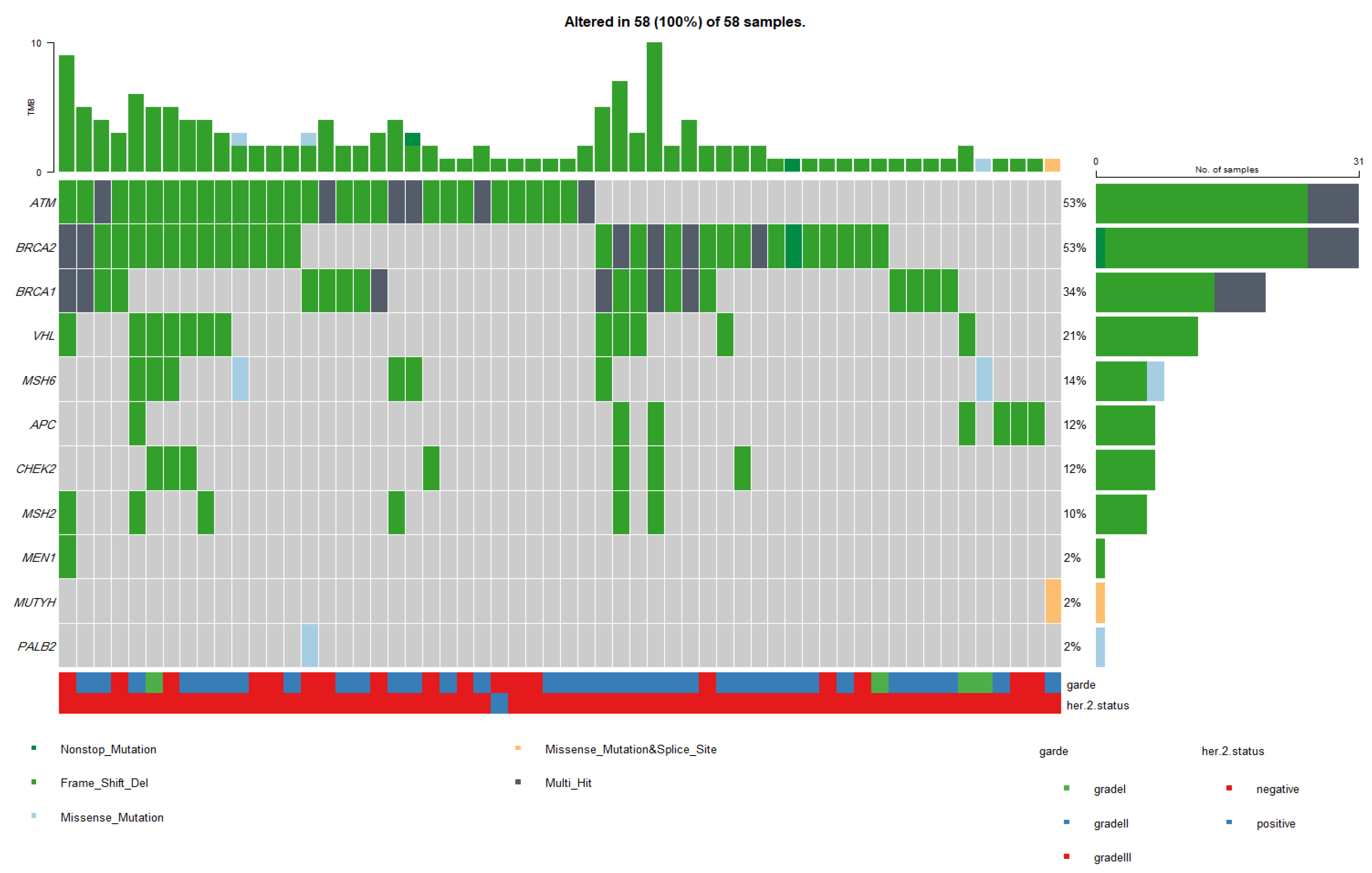
3.6. Patients with Pathogenic Germline Mutations in Single and Multiple Genes
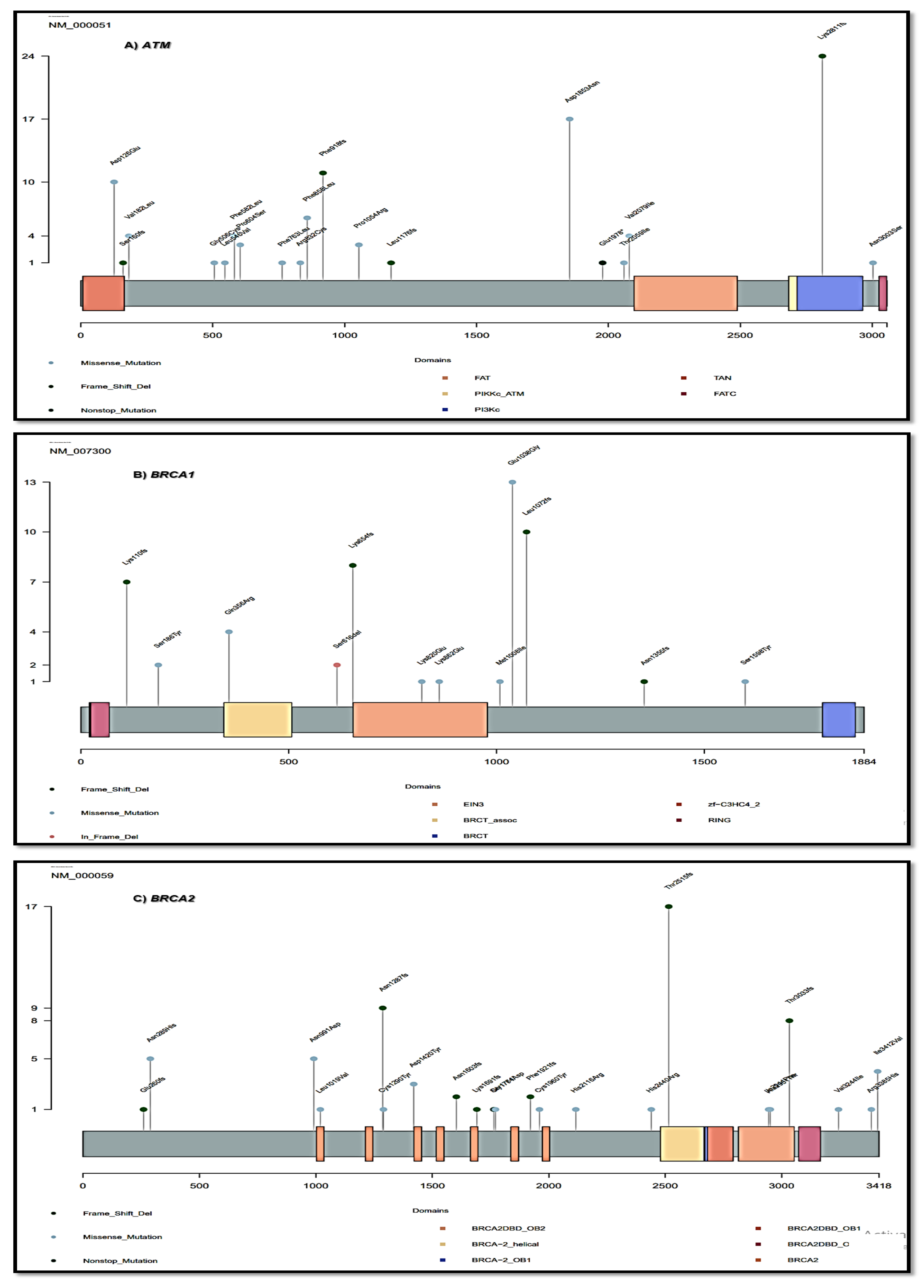
3.7. The Distribution of Pathogenic Gene Mutations in Exon Regions and Protein Domains
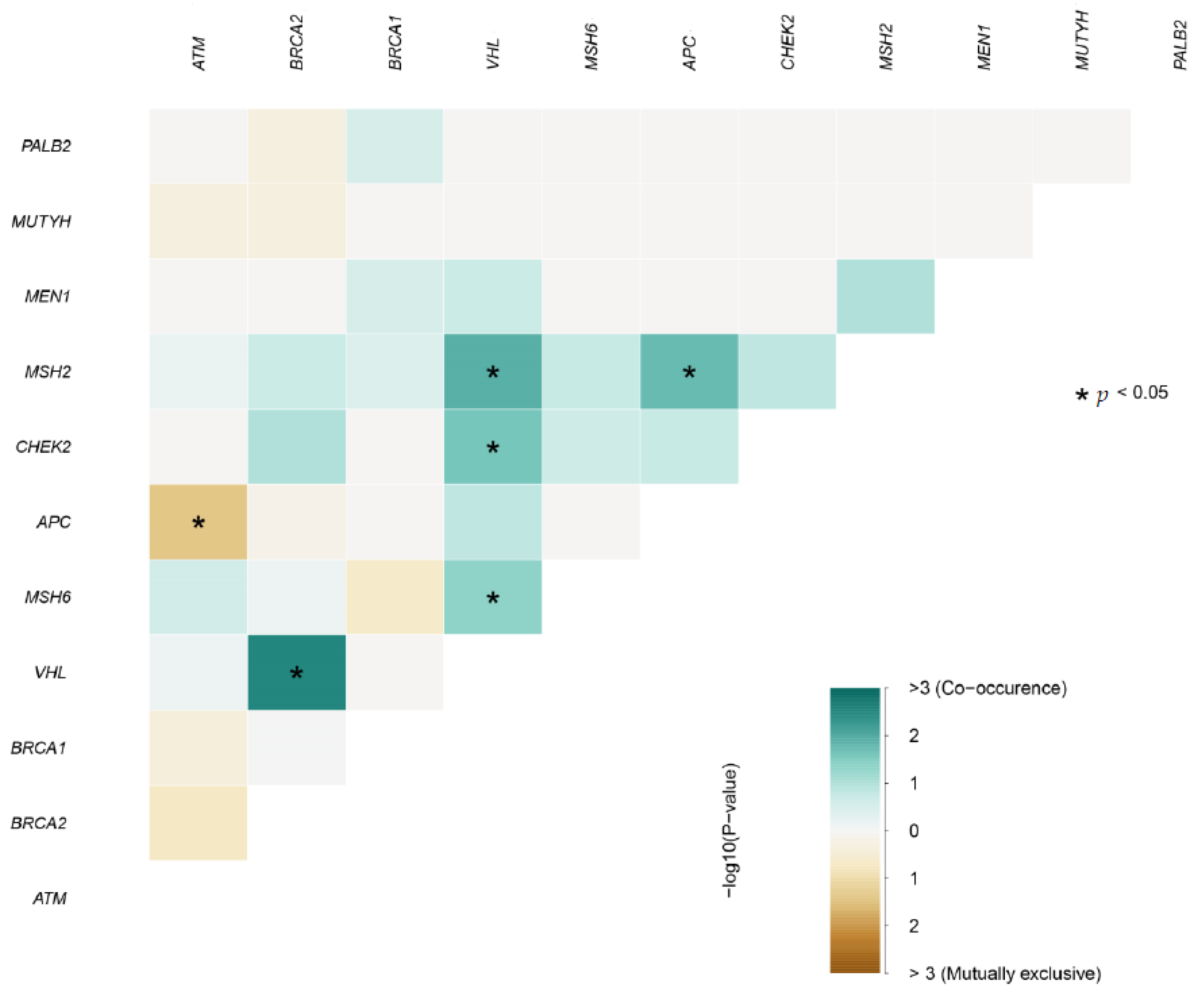
3.8. Mutually Exclusive and Co-Occurring Events between Gene Pairs with Deleterious Mutations
4. Discussion

Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Breast Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed on 24 July 2022).
- Ibrahim, A.S.; Khaled, H.M.; Mikhail, N.N.; Baraka, H.; Kamel, H. Cancer Incidence in Egypt: Results of the National Population-Based Cancer Registry Program. J. Cancer Epidemiol. 2014, 2014, 437971. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Spezia, M.; Huang, S.; Yuan, C.; Zeng, Z.; Zhang, L.; Ji, X.; Liu, W.; Huang, B.; Luo, W.; et al. Breast Cancer Development and Progression: Risk Factors, Cancer Stem Cells, Signaling Pathways, Genomics, and Molecular Pathogenesis. Genes Dis. 2018, 5, 77–106. [Google Scholar] [CrossRef]
- Bhushan, A.; Gonsalves, A.; Menon, J.U. Current State of Breast Cancer Diagnosis, Treatment, and Theranostics. Pharmaceutics 2021, 13, 723. [Google Scholar] [CrossRef]
- Yates, L.R.; Seoane, J.; Le Tourneau, C.; Siu, L.L.; Marais, R.; Michiels, S.; Soria, J.C.; Campbell, P.; Normanno, N.; Scarpa, A.; et al. The European Society for Medical Oncology (ESMO) Precision Medicine Glossary. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 30–35. [Google Scholar] [CrossRef]
- Mandelker, D.; Zhang, L.; Kemel, Y.; Stadler, Z.K.; Joseph, V.; Zehir, A.; Pradhan, N.; Arnold, A.; Walsh, M.F.; Li, Y.; et al. Mutation Detection in Patients With Advanced Cancer by Universal Sequencing of Cancer-Related Genes in Tumor and Normal DNA vs Guideline-Based Germline Testing. JAMA 2017, 318, 825–835. [Google Scholar] [CrossRef]
- Ratta, R.; Guida, A.; Scotté, F.; Neuzillet, Y.; Teillet, A.B.; Lebret, T.; Beuzeboc, P. PARP inhibitors as a new therapeutic option in metastatic prostate cancer: A systematic review. Prostate Cancer Prostatic Dis. 2020, 23, 549–560. [Google Scholar] [CrossRef]
- Scheuner, M.T.; Myrie, K.; Peredo, J.; Hoffman-Hogg, L.; Lundquist, M.; Guerra, S.L.; Ball, D. Integrating Germline Genetics Into Precision Oncology Practice in the Veterans Health Administration: Challenges and Opportunities. Federal practitioner: For the health care professionals of the VA, DoD, and PHS. Fed. Pract. 2020, 37, S82–S88. [Google Scholar]
- Gorodetska, I.; Kozeretska, I.; Dubrovska, A. BRCA Genes: The Role in Genome Stability, Cancer Stemness and Therapy Resistance. J. Cancer 2019, 10, 2109–2127. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, N.; Ryder, S.; Forbes, C.; Ross, J.; Quek, R.G. A Systematic Review of the International Prevalence of BRCA Mutation in Breast Cancer. Clin. Epidemiol. 2019, 11, 543–561. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Hu, L.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; Xu, Y.; Xie, Y. Germline Mutation in DNA-Repair Genes Is Associated with Poor Survival in BRCA1/2-Negative Breast Cancer Patients. Cancer Sci. 2019, 110, 3368–3374. [Google Scholar] [CrossRef] [Green Version]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.-C.; Lee, H.-B.; Yoo, T.-K.; Lee, E.-S.; Kim, R.N.; Park, B.; Yoon, K.-A.; Park, C.; Lee, E.S.; Moon, H.-G.; et al. Detection of Germline Mutations in Breast Cancer Patients with Clinical Features of Hereditary Cancer Syndrome Using a Multi-Gene Panel Test. Cancer Res. Treat. 2020, 52, 697–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbina-Jara, L.K.; Rojas-Martinez, A.; Martinez-Ledesma, E.; Aguilar, D.; Villarreal-Garza, C.; Ortiz-Lopez, R. Landscape of Germline Mutations in DNA Repair Genes for Breast Cancer in Latin America: Opportunities for PARP-Like Inhibitors and Immunotherapy. Genes 2019, 10, 786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Yang, J.; Li, L.; Cao, D.; Yu, M.; Shen, K. Germline and Somatic Mutations in Homologous Recombination Genes among Chinese Ovarian Cancer Patients Detected Using Next-Generation Sequencing. J. Gynecol. Oncol. 2017, 28, e39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espenschied, C.R.; LaDuca, H.; Li, S.; McFarland, R.; Gau, C.-L.; Hampel, H. Multigene Panel Testing Provides a New Perspective on Lynch Syndrome. J. Clin. Oncol. 2017, 35, 2568–2575. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA Repair, Genome Stability and Cancer: A Historical Perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Daly, M.B.; Pilarski, R.; Berry, M.; Buys, S.S.; Farmer, M.; Friedman, S.; Garber, J.E.; Kauff, N.D.; Khan, S.; Klein, C.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2017. J. Natl. Compr. Cancer Netw. 2017, 15, 9–20. [Google Scholar] [CrossRef]
- Li, J.; Li, H.; Makunin, I.; Thompson, B.A.; Tao, K.; Young, E.L.; Lopez, J.; Camp, N.J.; Tavtigian, S.V.; John, E.M.; et al. Panel Sequencing of 264 Candidate Susceptibility Genes and Segregation Analysis in a Cohort of Non-BRCA1, Non-BRCA2 Breast Cancer Families. Breast Cancer Res. Treat. 2017, 166, 937–949. [Google Scholar] [CrossRef]
- Kim, H.; Cho, D.-Y.; Choi, D.H.; Oh, M.; Shin, I.; Park, W.; Huh, S.J.; Nam, S.J.; Lee, J.E.; Kim, S.W. Frequency of Pathogenic Germline Mutation in CHEK2, PALB2, MRE11, and RAD50 in Patients at High Risk for Hereditary Breast Cancer. Breast Cancer Res. Treat. 2017, 161, 95–102. [Google Scholar] [CrossRef]
- Saied, M.; Elkaffash, D.; Fadl, R.; Haleem, R.; Refeat, A.; Ibrahim, I.; Tahoun, M.; Elkayal, A.; Tayae, E. Preliminary Results of Targeted Sequencing of BRCA1 and BRCA2 in a Cohort of Breast Cancer Families: New Insight into Pathogenic Variants in Patients and At-risk Relatives. Mol. Med. Rep. 2021, 24, 678. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Soliman, A.S.; Cui, J.; Ramadan, M.; Hablas, A.; Abouelhoda, M.; Hussien, N.; Ahmed, O.; Zekri, A.-R.N.; Seifeldin, I.A.; et al. Unique Features of Germline Variation in Five Egyptian Familial Breast Cancer Families Revealed by Exome Sequencing. PLoS ONE 2017, 12, e0167581. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the Effects of Coding Non-Synonymous Variants on Protein Function Using the SIFT Algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving Access to Variant Interpretations and Supporting Evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the Deleteriousness of Variants throughout the Human Genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Hyman, D.M.; Taylor, B.S.; Baselga, J. Implementing Genome-Driven Oncology. Cell 2017, 168, 584–599. [Google Scholar] [CrossRef] [Green Version]
- Rashid, M.U.; Muhammad, N.; Naeemi, H.; Khan, F.A.; Hassan, M.; Faisal, S.; Gull, S.; Amin, A.; Loya, A.; Hamann, U. Spectrum and Prevalence of BRCA1/2 Germline Mutations in Pakistani Breast Cancer Patients: Results from a Large Comprehensive Study. Hered. Cancer Clin. Pract. 2019, 17, 27. [Google Scholar] [CrossRef]
- Shah, P.D.; Nathanson, K.L. Application of Panel-Based Tests for Inherited Risk of Cancer. Annu. Rev. Genom. Hum. Genet. 2017, 18, 201–227. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.C.P.; Brett, M.; Lai, A.H.M.; Lee, S.-P.; Tan, E.-S.; Jamuar, S.S.; Ng, I.S.L.; Tan, E.-C. Next-Generation Sequencing Using a Pre-Designed Gene Panel for the Molecular Diagnosis of Congenital Disorders in Pediatric Patients. Hum. Genom. 2015, 9, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saudi Mendeliome Group. Comprehensive Gene Panels Provide Advantages over Clinical Exome Sequencing for Mendelian Diseases. Genome Biol. 2015, 16, 134. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, C.; Prasad, R.A.; Nfonsam, V.; Bernstei, H. DNA Damage, DNA Repair and Cancer. In New Research Directions in DNA Repair; InTechOpen: London, UK, 2013. [Google Scholar]
- Welcsh, P.L.; King, M.C. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum. Mol Genet. 2001, 10, 705–713. [Google Scholar] [CrossRef]
- Jalkh, N.; Chouery, E.; Haidar, Z.; Khater, C.; Atallah, D.; Ali, H.; Marafie, M.J.; Al-Mulla, M.R.; Al-Mulla, F.; Megarbane, A. Next-Generation Sequencing in Familial Breast Cancer Patients from Lebanon. BMC Med. Genom. 2017, 10, 8. [Google Scholar] [CrossRef] [Green Version]
- Al Hannan, F.; Keogh, M.B.; Taha, S.; Al Buainain, L. Characterization of BRCA1 and BRCA2 Genetic Variants in a Cohort of Bahraini Breast Cancer Patients Using Next-generation Sequencing. Mol. Genet. Genom. Med. 2019, 7, e00771. [Google Scholar] [CrossRef] [Green Version]
- Bu, R.; Siraj, A.K.; Al-Obaisi, K.A.S.; Beg, S.; Al Hazmi, M.; Ajarim, D.; Tulbah, A.; Al-Dayel, F.; Al-Kuraya, K.S. Identification of Novel BRCA Founder Mutations in Middle Eastern Breast Cancer Patients Using Capture and Sanger Sequencing Analysis. Int. J. Cancer 2016, 139, 1091–1097. [Google Scholar] [CrossRef] [Green Version]
- Bujassoum, S.M.; Bugrein, H.A.; Sulaiman, R. Al Genotype and Phenotype Correlation of Breast Cancer in BRCA Mutation Carriers and Non-Carriers. J. Cancer Sci. Ther. 2017, 9, 358–364. [Google Scholar] [CrossRef]
- Ben Ayed-Guerfali, D.; Ben Kridis-Rejab, W.; Ammous-Boukhris, N.; Ayadi, W.; Charfi, S.; Khanfir, A.; Sellami-Boudawara, T.; Frikha, M.; Daoud, J.; Mokdad-Gargouri, R. Novel and Recurrent BRCA1/BRCA2 Germline Mutations in Patients with Breast/Ovarian Cancer: A Series from the South of Tunisia. J. Transl. Med. 2021, 19, 108. [Google Scholar] [CrossRef]
- El Ansari, F.Z.; Jouali, F.; Marchoudi, N.; Bennani, M.M.; Ghailani, N.N.; Barakat, A.; Fekkak, J. Screening of BRCA1/2 Genes Mutations and Copy Number Variations in Patients with High Risk for Hereditary Breast and Ovarian Cancer Syndrome (HBOC). BMC Cancer 2020, 20, 747. [Google Scholar] [CrossRef]
- Mehemmai, C.; Cherbal, F.; Hamdi, Y.; Guedioura, A.; Benbrahim, W.; Bakour, R.; Abdelhak, S. BRCA1 and BRCA2 Germline Mutation Analysis in Hereditary Breast/Ovarian Cancer Families from the Aures Region (Eastern Algeria): First Report. Pathol. Oncol. Res. 2020, 26, 715–726. [Google Scholar] [CrossRef]
- Wong, E.S.Y.; Shekar, S.; Met-Domestici, M.; Chan, C.; Sze, M.; Yap, Y.S.; Rozen, S.G.; Tan, M.-H.; Ang, P.; Ngeow, J.; et al. Inherited Breast Cancer Predisposition in Asians: Multigene Panel Testing Outcomes from Singapore. NPJ Genom. Med. 2016, 1, 15003. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.R.; Devi, B.C.R.; Sung, H.; Guida, J.; Mucaki, E.J.; Xiao, Y.; Best, A.; Garland, L.; Xie, Y.; Hu, N.; et al. Prevalence and Spectrum of Germline Rare Variants in BRCA1/2 and PALB2 among Breast Cancer Cases in Sarawak, Malaysia. Breast Cancer Res. Treat. 2017, 165, 687–697. [Google Scholar] [CrossRef]
- ElBiad, O.; Laraqui, A.; El Boukhrissi, F.; Mounjid, C.; Lamsisi, M.; Bajjou, T.; Elannaz, H.; Lahlou, A.I.; Kouach, J.; Benchekroune, K.; et al. Prevalence of Specific and Recurrent/Founder Pathogenic Variants in BRCA Genes in Breast and Ovarian Cancer in North Africa. BMC Cancer 2022, 22, 208. [Google Scholar] [CrossRef]
- Loza, P.; Irmejs, A.; Daneberga, Z.; Miklasevics, E.; Berga-Svitina, E.; Subatniece, S.; Maksimenko, J.; Trofimovics, G.; Tauvena, E.; Ukleikins, S.; et al. A Novel Frequent BRCA1 Recurrent Variant c.5117G > A (p.Gly1206Glu) Identified after 20 Years of BRCA1/2 Research in the Baltic Region: Cohort Study and Literature Review. Hered. Cancer Clin. Pract. 2021, 19, 11. [Google Scholar] [CrossRef]
- Bahsi, T.; Erdem, H.B. Spectrum of BRCA1/BRCA2 Variants in 1419 Turkish Breast and Ovarian Cancer Patients: A Single Center Study. Turk. J. Biochem. 2020, 45, 83–90. [Google Scholar] [CrossRef]
- Palmero, E.I.; Carraro, D.M.; Alemar, B.; Moreira, M.A.M.; Ribeiro-dos-Santos, Â.; Abe-Sandes, K.; Galvão, H.C.R.; Reis, R.M.; de Pádua Souza, C.; Campacci, N.; et al. The Germline Mutational Landscape of BRCA1 and BRCA2 in Brazil. Sci. Rep. 2018, 8, 9188. [Google Scholar] [CrossRef] [Green Version]
- Novaković, S.; Milatović, M.; Cerkovnik, P.; Stegel, V.; Krajc, M.; Hočevar, M.; Žgajnar, J.; Vakselj, A. Novel BRCA1 and BRCA2 Pathogenic Mutations in Slovene Hereditary Breast and Ovarian Cancer Families. Int. J. Oncol. 2012, 41, 1619–1627. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, W.; Shi, Y.; Huang, Y.; Sun, T.; Tang, L.; Lu, Q.; Lei, Q.; Liao, N.; Jin, F.; et al. Germline Mutation Landscape of Chinese Patients with Familial Breast/Ovarian Cancer in a Panel of 22 Susceptibility Genes. Cancer Med. 2019, 8, 2074–2084. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Meng, H.; Yao, L.; Lv, M.; Bai, J.; Zhang, J.; Wang, L.; Ouyang, T.; Li, J.; Wang, T.; et al. Germline Mutations in Cancer Susceptibility Genes in a Large Series of Unselected Breast Cancer Patients. Clin. Cancer Res. 2017, 23, 6113–6119. [Google Scholar] [CrossRef] [Green Version]
- Economopoulou, P.; Dimitriadis, G.; Psyrri, A. Beyond BRCA: New Hereditary Breast Cancer Susceptibility Genes. Cancer Treat. Rev. 2015, 41, 1–8. [Google Scholar] [CrossRef]
- Stolarova, L.; Kleiblova, P.; Janatova, M.; Soukupova, J.; Zemankova, P.; Macurek, L.; Kleibl, Z. CHEK2 Germline Variants in Cancer Predisposition: Stalemate Rather than Checkmate. Cells 2020, 9, 2675. [Google Scholar] [CrossRef]
- Ranjha, L.; Howard, S.M.; Cejka, P. Main Steps in DNA Double-Strand Break Repair: An Introduction to Homologous Recombination and Related Processes. Chromosoma 2018, 127, 187–214. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Keijzers, G.; Rasmussen, L.J. DNA Mismatch Repair and Its Many Roles in Eukaryotic Cells. Mutat. Res. Rev. Mutat. Res. 2017, 773, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Tulay, P.; Sengupta, S.B. MicroRNA Expression and Its Association with DNA Repair in Preimplantation Embryos. J. Reprod. Dev. 2016, 62, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- U.S. Food and Drug Administration. FDA Approves Olaparib for Germline BRCA-Mutated Metastatic Breast Cancer. 2022. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-germline-brca-mutated-metastatic-breast-cancer (accessed on 3 September 2022).
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Petermann, E.; Yates, E.; Brown, J.; Lau, A.; Stankovic, T. Synthetic Lethality in Chronic Lymphocytic Leukaemia with DNA Damage Response Defects by Targeting the ATR Pathway. Lancet 2015, 385, S58. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-Art Strategies for Targeting the DNA Damage Response in Cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Chae, Y.K.; Anker, J.F.; Bais, P.; Namburi, S.; Giles, F.J.; Chuang, J.H. Mutations in DNA repair genes are associated with increased neo-antigen load and activated T cell infiltration in lung adenocarcinoma. Oncotarget 2018, 9, 7949–7960. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, K.N.; Wubbenhorst, B.; D’Andrea, K.; Garman, B.; Long, J.M.; Powers, J.; Rathbun, K.; Stopfer, J.E.; Zhu, J.; Bradbury, A.R.; et al. Prevalence of Mutations in a Panel of Breast Cancer Susceptibility Genes in BRCA1/2-Negative Patients with Early-Onset Breast Cancer. Genet. Med. 2015, 17, 630–638. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Meeks, H.; Feng, B.-J.; Healey, S.; Thorne, H.; Makunin, I.; Ellis, J.; Campbell, I.; Southey, M.; Mitchell, G.; et al. Targeted Massively Parallel Sequencing of a Panel of Putative Breast Cancer Susceptibility Genes in a Large Cohort of Multiple-Case Breast and Ovarian Cancer Families. J. Med. Genet. 2016, 53, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Thompson, E.R.; Rowley, S.M.; Li, N.; McInerny, S.; Devereux, L.; Wong-Brown, M.W.; Trainer, A.H.; Mitchell, G.; Scott, R.J.; James, P.A.; et al. Panel Testing for Familial Breast Cancer: Calibrating the Tension Between Research and Clinical Care. J. Clin. Oncol. 2016, 34, 1455–1459. [Google Scholar] [CrossRef] [Green Version]
- Lang, G.-T.; Shi, J.-X.; Huang, L.; Cao, A.-Y.; Zhang, C.-H.; Song, C.-G.; Zhuang, Z.-G.; Hu, X.; Huang, W.; Shao, Z.-M. Multiple Cancer Susceptible Genes Sequencing in BRCA-Negative Breast Cancer with High Hereditary Risk. Ann. Transl. Med. 2020, 8, 1417. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Total (%) 101 (100) | P/LP Mutation Carriers | P/LP Mutation Non-Carriers | p-Value |
|---|---|---|---|---|
| Number of patients | 101 (100) | 58 | 43 | |
| BC Grade | ||||
| I | 7 (6.9) | 4 | 3 | |
| II | 67 (66.3) | 36 | 31 | 0.52 |
| III | 27 (26.7) | 18 | 9 | |
| Tumor type | ||||
| Invasive ductal carcinoma | 94 (93.06) | 52 | 42 | |
| Invasive tubular carcinoma | 2 (1.98) | 2 | 0 | 0.41 |
| Invasive lobular carcinoma | 4 (3.96) | 3 | 1 | |
| micropapillary carcinoma | 1 (0.99) | 1 | 0 | |
| Age at diagnosis | ||||
| BC diagnosis at ≤40 years | 72 (71.3) | 41 | 31 | 0.88 |
| BC diagnosis at >40 years | 29 (28.7) | 17 | 12 | |
| BRCA1/2 status | ||||
| BRCA1/2 positive | 40 (39.6) | 40 * | 0 | <0.001 ** |
| BRCA1/2 negative | 61 (60.4) | 18 * | 43 |
| Gene | Position | dbSNP | Frequency | Type | Clinical Significance | Exon | HGVS.c | HGVS.p | Grade 1 | Grade 2 | Grade 3 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ATM | Chr11:10821647 | rs587782558 | 24 | Indel | PV | 57 | c.8431_8432delAA | p.Lys2811fs | 1 | 12 | 11 |
| Chr11:108139249 | rs786202608 | 11 | Indel | PV | 18 | c.2754delT | p.Phe918fs | - | 8 | 3 | |
| Chr11:108151842 | rs730881302 | 1 | Indel | PV | 24 | c.3526delC | p.Leu1176fs | - | 1 | - | |
| Chr11:108106541 | rs587780624 | 1 | Indel | LPV | 5 | c.478_482delTCTCA | p.Ser160fs | - | - | 1 | |
| Chr11:108183151 | rs587779852 | 1 | SNP (Stop gain) | PV | 40 | c.5932G > T | p.Glu1978 * | - | 1 | - | |
| BRCA2 | Chr13:32930667 | rs80359657 | 17 | Indel | PV | 15 | c.7543delA | p.Thr2515fs | 2 | 12 | 3 |
| Chr13:32912345 | rs80359406 | 9 | Indel | PV | 11 | c.3860delA | p.Asn1287fs | - | 7 | 2 | |
| Chr13:32954022 | rs397507419 | 8 | Indel | PV | 23 | c.9097delA | p.Thr3033fs | - | 4 | 4 | |
| Chr13:32913558 | rs80359479 | 1 | Indel | PV | 11 | c.5073delA | p.Lys1691fs | - | 1 | - | |
| Chr13:32913295 | rs80359466 | 2 | Indel | PV | 11 | c.4808delA | p.Asn1603fs | - | 1 | 1 | |
| Chr13:32913783 | rs397507778 | 1 | SNP (Stop gain) | PV | 11 | c.5291C > G | p.Ser1764 * | - | 1 | - | |
| Chr13:32914250 | rs80359534 | 2 | Indel | PV | 11 | c.5763delT | p.Phe1921fs | - | 2 | - | |
| Chr13:32905146 | rs75096777 | 1 | Indel | PV | 9 | c.774_775delAA | p.Glu260fs | - | 1 | - | |
| BRCA1 | Chr17:41244333 | rs80357923 | 10 | Indel | PV | 10 | c.3214delC | p.Leu1072fs | - | 9 | 1 |
| Chr17:41245586 | rs80357522 | 8 | Indel | PV | 10 | c.1961delA | p.Lys654fs | - | 4 | 4 | |
| Chr17:41256250 | rs80357604 | 7 | Indel | PV | 5 | c.329delA | p.Lys110fs | - | 5 | 2 | |
| Chr17:41243479 | rs80357508 | 1 | Indel | PV | 10 | c.4065_4068delTCAA | p.Asn1355fs | - | - | 1 | |
| VHL | Chr3:10188296 | rs869025653 | 12 | Indel | PV | 2 | c.444delT | p.Phe148fs | 2 | 8 | 2 |
| MSH6 | Chr2:48030691 | rs267608092 | 6 | Indel | PV | 5 | c.3312delT | p.Phe1104fs | 1 | 4 | 1 |
| Chr2:48025764 | rs1800937 | 2 | SNP | PV | 3 | c.642C > A | p.Tyr214Ter | 1 | 1 | - | |
| APC | Chr5:112175100 | rs587783033 | 6 | Indel | PV | 15 | c.3814delT | p.Ser1272fs | 1 | 3 | 2 |
| Chr5:112173393 | rs587783030 | 1 | Indel | PV | 15 | c.2107delG | p.Ala703fs | - | 1 | - | |
| CHEK2 | Chr22:29099524 | rs772683219 | 7 | Indel | PV | 9 | c.1005delT | p.Phe335fs | 1 | 4 | 2 |
| MSH2 | Chr2:47709924 | rs63750084 | 6 | Indel | PV | 16 | c.2647delA | p.Ile883fs | - | 5 | 1 |
| MEN1 | Chr11:64572092 | rs794728642 | 1 | Indel | PV | 10 | c.1561delC | p.Arg521fs | - | - | 1 |
| PALB2 | Chr16:23646857 | rs45494092 | 1 | SNP | PV | 4 | c.1010T > A | p.Leu337Ter | - | - | 1 |
| MUTYH | Chr1:45797228 | rs36053993 | 1 | SNP | LPV | 13 | c.1187G > A | p.Gly396Asp | - | 1 | - |
| DNA Repair Genes | Mutation Cases (Out of 101) | Prevalence (%) | Grade I (%) | Grade II (%) | Grade III (%) | p-Value | ||
|---|---|---|---|---|---|---|---|---|
| Double strand repair | HRR | BRCA1 | 20 | 19.8 | 14 (70) | 6 (30) | 0.42 | |
| BRCA2 | 31 | 30.7 | 2 (6.5) | 21 (67.7) | 8 (25.8) | |||
| PALB2 | 1 | 0.99 | 1 (100) | |||||
| Single strand repair | MMR | MSH2 | 6 | 5.6 | 5 (83.3) | 1 (16.7) | 0.69 | |
| MSH6 | 8 | 7.9 | 2 (25) | 5 (62.5) | 1 (12.5) | |||
| BER | MUTYH | 1 | 0.99 | 1 (100) | ||||
| Checkpoint | ATM | 31 | 30.7 | 1 (3.22) | 17 (54.84) | 13 (41.94) | 0.45 | |
| CHEK2 | 7 | 6.9 | 1 (14.29) | 4 (57.14) | 2 (28.57) | 0.42 | ||
| Gene | Affected Transcript | Patient No. (Alteration in 1 Gene) | Patient No. (Alteration in 2 Genes) | Patient No. (Alteration in 3 Genes) | Patient No. (Alteration in 4 Genes) | Patient No. (Alteration in 5 Genes) | Patient No. (Alteration in 6 Genes) |
| BRCA1 | c.1961delA | Sample 6 * | Sample 1 * Sample 22 Sample 36 | Sample 86 * | Sample 70 * | Sample 87 * | |
| Sample 34 | |||||||
| c.329delA | Sample 30 * | Sample 82 | Sample 86 * | Sample 70 * | Sample 87 * Sample 90 | ||
| Sample 15 | |||||||
| c.4065_4068delTCAA | Sample 30 * | ||||||
| c.3214delC | Sample 16 Sample 17 Sample 7 Sample 51 | Sample 6 * Sample 21 Sample 25 Sample 28 | Sample 1 * Sample 32 | ||||
| BRCA 2 | c.7543delA | Sample 72 Sample 97 | Sample 6 * Sample 15 Sample 78 Sample 98 | Sample 82 Sample 71 | Sample 86 Sample 79 Sample 84 | Sample 70 * Sample 55 Sample 89 | Sample 87 * Sample 90 * Sample 66 |
| c.3860delA | Sample 20 * Sample 42 | Sample 1 * Sample 32 Sample 4 Sample 36 | Sample 70 * | Sample 90 * Sample 87 * | |||
| c.9097delA | Sample 20 * Sample 41 Sample 39 | Sample 6 * Sample 34 Sample 29 Sample 43 Sample 50 | |||||
| c.5073delA | Sample 70 * | ||||||
| c.4808delA | Sample 70 * | Sample 87 * | |||||
| c.5291C > G | Sample 33 | ||||||
| c.5763delT | Sample 1 * | Sample 70 * | |||||
| c.774_775delAA | Sample 8 | ||||||
| ATM | c.8431_8432delAA | Sample 19 Sample 53 Sample 35 Sample 96 * Sample 104 * | Sample 21 * Sample 30 Sample 25 Sample 28 Sample 29 Sample 43 Sample 94 | Sample 1 Sample 32 * Sample 99 * Sample 22 Sample 71 Sample 4 Sample 36 | Sample 79 Sample 84 | Sample 55 Sample 89 | Sample 87 |
| c.2754delT | Sample 56 | Sample 21 * Sample 60 * | Sample 32 * Sample 99 * | Sample 66 | |||
| Sample 64 | |||||||
| Sample 77 Sample 96 * | |||||||
| Sample 101 Sample 104 * | |||||||
| c.478_482delTCTCA | Sample 21 * | ||||||
| c.3526delC | Sample 50 | ||||||
| c.5932G > T | Sample 60 * | ||||||
| VHL | c.444delT | Sample 83 Sample 98 | Sample 82 Sample 71 | Sample 86 Sample 79 Sample 84 | Sample 55 Sample 89 | Sample 87 Sample 90 Sample 66 | |
| MSH6 | c.642C > A | Sample 38 | Sample 4 | ||||
| c.3312delT | Sample 60 | Sample 99 | Sample 86 | Sample 55 Sample 89 | Sample 66 | ||
| APC | c.3814delT | Sample 58 Sample 65 | Sample 83 | Sample 70 | Sample 90 Sample 66 | ||
| c.2107delG | Sample 48 | ||||||
| CHEK2 | c.1005delT | Sample 78 Sample 94 | Sample 84 | Sample 70 Sample 55 Sample 89 | Sample 90 | ||
| MSH2 | c.2647delA | Sample 99 | Sample 79 | Sample 70 | Sample 87 Sample 90 Sample 66 | ||
| MEN1 | c.1561delC | Sample 87 | |||||
| PALB2 | c.1010T > A | Sample 22 | |||||
| MUTYH | c.1187G > A | Sample 102 |
| dbSNP | Gene Name | Clinical Significance | dbSNP | Gene Name | Clinical Significance |
|---|---|---|---|---|---|
| rs146297864 | NEK2 | VUS | rs730881396 | AXIN2 | VUS |
| rs1799939 | RET | Conflicting interpretations of pathogenicity | rs28997569 | BRIP1 | Conflicting interpretations of pathogenicity |
| rs607969 | MEN1 | Conflicting interpretations of pathogenicity | rs367696886 | XRCC2 | VUS |
| rs2229022 | ATM | Conflicting interpretations of pathogenicity | rs372305287 | APC | Conflicting interpretations of pathogenicity |
| rs144636562 | ATM | VUS | rs371264852 | STK11 | Conflicting interpretations of pathogenicity |
| rs770406711 | APC | VUS | rs373226409 | MSH2 | Conflicting interpretations of pathogenicity |
| rs138743097 | BRIP1 | VUS | rs4988345 | BRIP1 | Conflicting interpretations of pathogenicity |
| rs138749920 | RAD50 | VUS | rs587782683 | MUTYH | VUS |
| rs138933660 | APC | Conflicting interpretations of pathogenicity | rs62625284 | PALB2 | Conflicting interpretations of pathogenicity |
| rs777004819 | MER11A | VUS | rs786203658 | NF1 | VUS |
| rs141142822 | ERBB2 | VUS | rs751431238 | MSH2 | VUS |
| rs145415033 | MER11A | VUS | rs759105985 | PALID | VUS |
| rs149342980 | MUTYH | VUS | rs761673463 | BARD1 | VUS |
| rs149815077 | GEN1 | VUS | rs180727534 | SYNE1 | VUS |
| rs189059377 | BMPR1A | VUS | rs201707558 | PALLD | VUS |
| rs273901741 | BRCA1 | Conflicting interpretations of pathogenicity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nassar, A.; Zekri, A.-R.N.; Kamel, M.M.; Elberry, M.H.; Lotfy, M.M.; Seadawy, M.G.; Hassan, Z.K.; Soliman, H.K.; Lymona, A.M.; Youssef, A.S.E.-D. Frequency of Pathogenic Germline Mutations in Early and Late Onset Familial Breast Cancer Patients Using Multi-Gene Panel Sequencing: An Egyptian Study. Genes 2023, 14, 106. https://doi.org/10.3390/genes14010106
Nassar A, Zekri A-RN, Kamel MM, Elberry MH, Lotfy MM, Seadawy MG, Hassan ZK, Soliman HK, Lymona AM, Youssef ASE-D. Frequency of Pathogenic Germline Mutations in Early and Late Onset Familial Breast Cancer Patients Using Multi-Gene Panel Sequencing: An Egyptian Study. Genes. 2023; 14(1):106. https://doi.org/10.3390/genes14010106
Chicago/Turabian StyleNassar, Auhood, Abdel-Rahman N. Zekri, Mahmoud M. Kamel, Mostafa H. Elberry, Mai M. Lotfy, Mohamed G. Seadawy, Zeinab K. Hassan, Hany K. Soliman, Ahmed M. Lymona, and Amira Salah El-Din Youssef. 2023. "Frequency of Pathogenic Germline Mutations in Early and Late Onset Familial Breast Cancer Patients Using Multi-Gene Panel Sequencing: An Egyptian Study" Genes 14, no. 1: 106. https://doi.org/10.3390/genes14010106