
Novel Exon-Skipping Therapeutic Approach for the DMD Gene Based on Asymptomatic Deletions of Exon 49

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Ethical Statement
2.3. Polymerase Chain Reaction (PCR) and Sequencing
2.4. Cell Culture
2.5. Differentiation into Myotubes
2.6. Antisense Oligonucleotides (AON)
2.7. Transfection of Myotubes with AON
2.8. RNA Extraction and Reverse Transcription Polymerase Chain Reaction (RT-PCR)
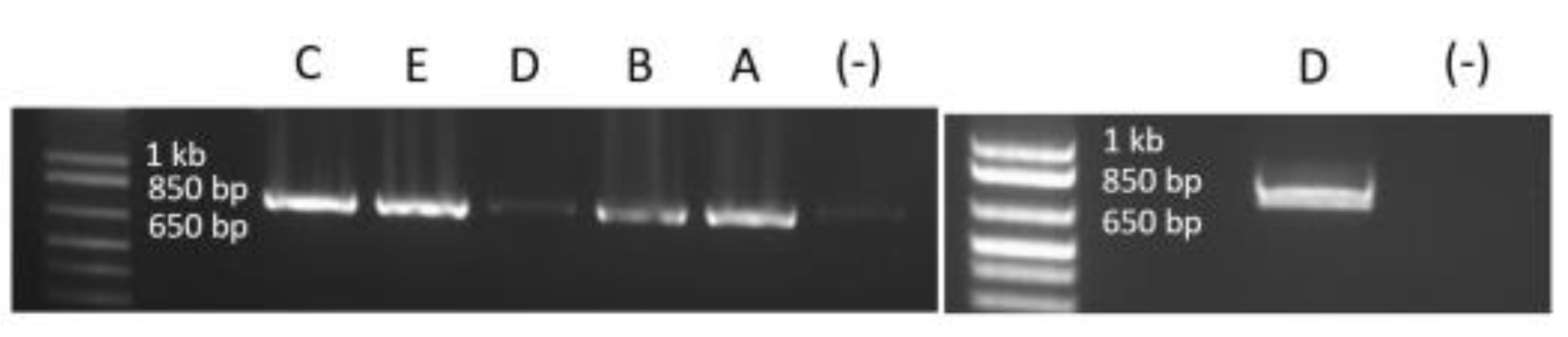
3. Results
3.1. Clinical Observations
3.2. Genomic Position of the Observed Deletion
3.3. In Vitro Application of Exon 49 Skipping in WT and Mutated Myotubes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-Based Path to Newborn Screening for Duchenne Muscular Dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef]
- Nigro, G.; Comi, L.I.; Politano, L.; Bain, R.J. The Incidence and Evolution of Cardiomyopathy in Duchenne Muscular Dystrophy. Int. J. Cardiol. 1990, 26, 271–277. [Google Scholar] [CrossRef]
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. In Comprehensive Physiology; Terjung, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 1223–1239. ISBN 978-0-470-65071-4. [Google Scholar]
- Flanigan, K.M.; Dunn, D.M.; von Niederhausern, A.; Soltanzadeh, P.; Gappmaier, E.; Howard, M.T.; Sampson, J.B.; Mendell, J.R.; Wall, C.; King, W.M.; et al. Mutational Spectrum of DMD Mutations in Dystrophinopathy Patients: Application of Modern Diagnostic Techniques to a Large Cohort. Hum. Mutat. 2009, 30, 1657–1666. [Google Scholar] [CrossRef] [Green Version]
- Tuffery-Giraud, S.; Béroud, C.; Leturcq, F.; Yaou, R.B.; Hamroun, D.; Michel-Calemard, L.; Moizard, M.-P.; Bernard, R.; Cossée, M.; Boisseau, P.; et al. Genotype-Phenotype Analysis in 2405 Patients with a Dystrophinopathy Using the UMD-DMD Database: A Model of Nationwide Knowledgebase. Hum. Mutat. 2009, 30, 934–945. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Van Deutekom, J.C.T.; Fokkema, I.F.; Van Ommen, G.-J.B.; Den Dunnen, J.T. Entries in the Leiden Duchenne Muscular Dystrophy Mutation Database: An Overview of Mutation Types and Paradoxical Cases That Confirm the Reading-Frame Rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Waldrop, M.A.; Yaou, R.B.; Lucas, K.K.; Martin, A.S.; O’Rourke, E.; Ferlini, A.; Muntoni, F.; Leturcq, F.; Tuffery-Giraud, S.; Weiss, R.B.; et al. Clinical Phenotypes of DMD Exon 51 Skip 3 Equivalent Deletions: A Systematic Review. J. Neuromuscul. Dis. 2022, 7, 217–229. [Google Scholar] [CrossRef] [PubMed]
- van den Bergen, J.C.; Hiller, M.; Böhringer, S.; Vijfhuizen, L.; Ginjaar, H.B.; Chaouch, A.; Bushby, K.; Straub, V.; Scoto, M.; Cirak, S.; et al. Validation of Genetic Modifiers for Duchenne Muscular Dystrophy: A Multicentre Study Assessing SPP1 and LTBP4 Variants. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1060–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, R.B.; Vieland, V.J.; Dunn, D.M.; Kaminoh, Y.; Flanigan, K.M. Long-Range Genomic Regulators of THBS1 and LTBP4 Modify Disease Severity in Duchenne Muscular Dystrophy. Ann. Neurol. 2019, 20, 234–245. [Google Scholar] [CrossRef] [PubMed]
- CINRG Investigators; Spitali, P.; Zaharieva, I.; Bohringer, S.; Hiller, M.; Chaouch, A.; Roos, A.; Scotton, C.; Claustres, M.; Bello, L.; et al. TCTEX1D1 Is a Genetic Modifier of Disease Progression in Duchenne Muscular Dystrophy. Eur. J. Hum. Genet. 2020, 28, 815–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echevarría, L.; Aupy, P.; Goyenvalle, A. Exon-Skipping Advances for Duchenne Muscular Dystrophy. Hum. Mol. Genet. 2018, 27, R163–R172. [Google Scholar] [CrossRef] [Green Version]
- Aartsma-Rus, A.; Fokkema, I.; Verschuuren, J.; Ginjaar, I.; van Deutekom, J.; van Ommen, G.-J.; den Dunnen, J.T. Theoretic Applicability of Antisense-Mediated Exon Skipping for Duchenne Muscular Dystrophy Mutations. Hum. Mutat. 2009, 30, 293–299. [Google Scholar] [CrossRef]
- Dunckley, M.G.; Manoharan, M.; Villiet, P.; Eperon, I.C.; Dickson, G. Modification of Splicing in the Dystrophin Gene in Cultured Mdx Muscle Cells by Antisense Oligoribonucleotides. Hum. Mol. Genet. 1998, 7, 1083–1090. [Google Scholar] [CrossRef]
- Charleston, J.S.; Schnell, F.J.; Dworzak, J.; Donoghue, C.; Lewis, S.; Chen, L.; Young, G.D.; Milici, A.J.; Voss, J.; DeAlwis, U.; et al. Eteplirsen Treatment for Duchenne Muscular Dystrophy: Exon Skipping and Dystrophin Production. Neurology 2018, 90, e2146–e2154. [Google Scholar] [CrossRef]
- Wagner, K.R.; Kuntz, N.L.; Koenig, E.; East, L.; Upadhyay, S.; Han, B.; Shieh, P.B. Safety, Tolerability, and Pharmacokinetics of Casimersen in Patients with D Uchenne Muscular Dystrophy Amenable to Exon 45 Skipping: A Randomized, Double-blind, Placebo-controlled, Dose-titration Trial. Muscle Nerve 2021, 64, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Clemens, P.R.; Rao, V.K.; Connolly, A.M.; Harper, A.D.; Mah, J.K.; Smith, E.C.; McDonald, C.M.; Zaidman, C.M.; Morgenroth, L.P.; Osaki, H.; et al. Safety, Tolerability, and Efficacy of Viltolarsen in Boys With Duchenne Muscular Dystrophy Amenable to Exon 53 Skipping: A Phase 2 Randomized Clinical Trial. JAMA Neurol. 2020, 77, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the Treatment of Duchenne Muscular Dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef]
- Mital, A.; Kumari, D.; Gupta, M.; Goyle, S. Molecular Characterisation of Duchenne Muscular Dystrophy and Phenotypic Correlation. J. Neurol. Sci. 1998, 157, 179–186. [Google Scholar] [CrossRef]
- Banerjee, M.; Verma, I.C. Are There Ethnic Differences in Deletions in the Dystrophin Gene? Am. J. Med. Genet. 1997, 68, 152–157. [Google Scholar] [CrossRef]
- Zamani, G.R.; Karami, F.; Mehdizadeh, M.; Movafagh, A.; Nilipour, Y.; Zamani, M. Analysis of Dystrophin Gene in Iranian Duchenne and Becker Muscular Dystrophies Patients and Identification of a Novel Mutation. Neurol. Sci. 2015, 36, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Florencia, G.; Verónica, F.; Viviana, D.; Irene, S. Dystrophin Deletions and Cognitive Impairment in Duchenne/Becker Muscular Dystrophy. Neurol. Res. 2004, 26, 83–87. [Google Scholar] [CrossRef]
- Deepha, S.; Vengalil, S.; Preethish-Kumar, V.; Polavarapu, K.; Nalini, A.; Gayathri, N.; Purushottam, M. MLPA Identification of Dystrophin Mutations and in Silico Evaluation of the Predicted Protein in Dystrophinopathy Cases from India. BMC Med. Genet. 2017, 18, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, U.; Lee, S.-A.; Choi, W.A.; Kang, S.-W.; Seo, G.H.; Lee, J.H.; Park, G.; Lee, S.; Choi, Y.-C.; Park, H.J. Clinical and Genetic Spectra in Patients with Dystrophinopathy in Korea: A Single-Center Study. PLoS ONE 2021, 16, e0255011. [Google Scholar] [CrossRef] [PubMed]
- Marey, I.; Ben Yaou, R.; Deburgrave, N.; Vasson, A.; Nectoux, J.; Leturcq, F.; Eymard, B.; Laforet, P.; Behin, A.; Stojkovic, T.; et al. Non Random Distribution of DMD Deletion Breakpoints and Implication of Double Strand Breaks Repair and Replication Error Repair Mechanisms. J. Neuromuscul. Dis. 2016, 3, 227–245. [Google Scholar] [CrossRef]
- Brison, N.; Storms, J.; Villela, D.; Claeys, K.G.; Dehaspe, L.; de Ravel, T.; De Waele, L.; Goemans, N.; Legius, E.; Peeters, H.; et al. Maternal Copy-Number Variations in the DMD Gene as Secondary Findings in Noninvasive Prenatal Screening. Genet. Med. 2019, 21, 2774–2780. [Google Scholar] [CrossRef]
- Koenig, M.; Kunkel, L.M. Detailed Analysis of the Repeat Domain of Dystrophin Reveals Four Potential Hinge Segments That May Confer Flexibility. J. Biol. Chem. 1990, 265, 4560–4566. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abaji, M.; Gorokhova, S.; Da Silva, N.; Busa, T.; Grelet, M.; Missirian, C.; Sigaudy, S.; Philip, N.; Leturcq, F.; Lévy, N.; et al. Novel Exon-Skipping Therapeutic Approach for the DMD Gene Based on Asymptomatic Deletions of Exon 49. Genes 2022, 13, 1277. https://doi.org/10.3390/genes13071277
Abaji M, Gorokhova S, Da Silva N, Busa T, Grelet M, Missirian C, Sigaudy S, Philip N, Leturcq F, Lévy N, et al. Novel Exon-Skipping Therapeutic Approach for the DMD Gene Based on Asymptomatic Deletions of Exon 49. Genes. 2022; 13(7):1277. https://doi.org/10.3390/genes13071277
Chicago/Turabian StyleAbaji, Mario, Svetlana Gorokhova, Nathalie Da Silva, Tiffany Busa, Maude Grelet, Chantal Missirian, Sabine Sigaudy, Nicole Philip, France Leturcq, Nicolas Lévy, and et al. 2022. "Novel Exon-Skipping Therapeutic Approach for the DMD Gene Based on Asymptomatic Deletions of Exon 49" Genes 13, no. 7: 1277. https://doi.org/10.3390/genes13071277