
Co-Expression Analysis Reveals Differential Expression of Homologous Genes Associated with Specific Terpenoid Biosynthesis in Rehmannia glutinosa


Abstract
:1. Introduction
2. Results
2.1. Construction of Unigene Set and Functional Annotations
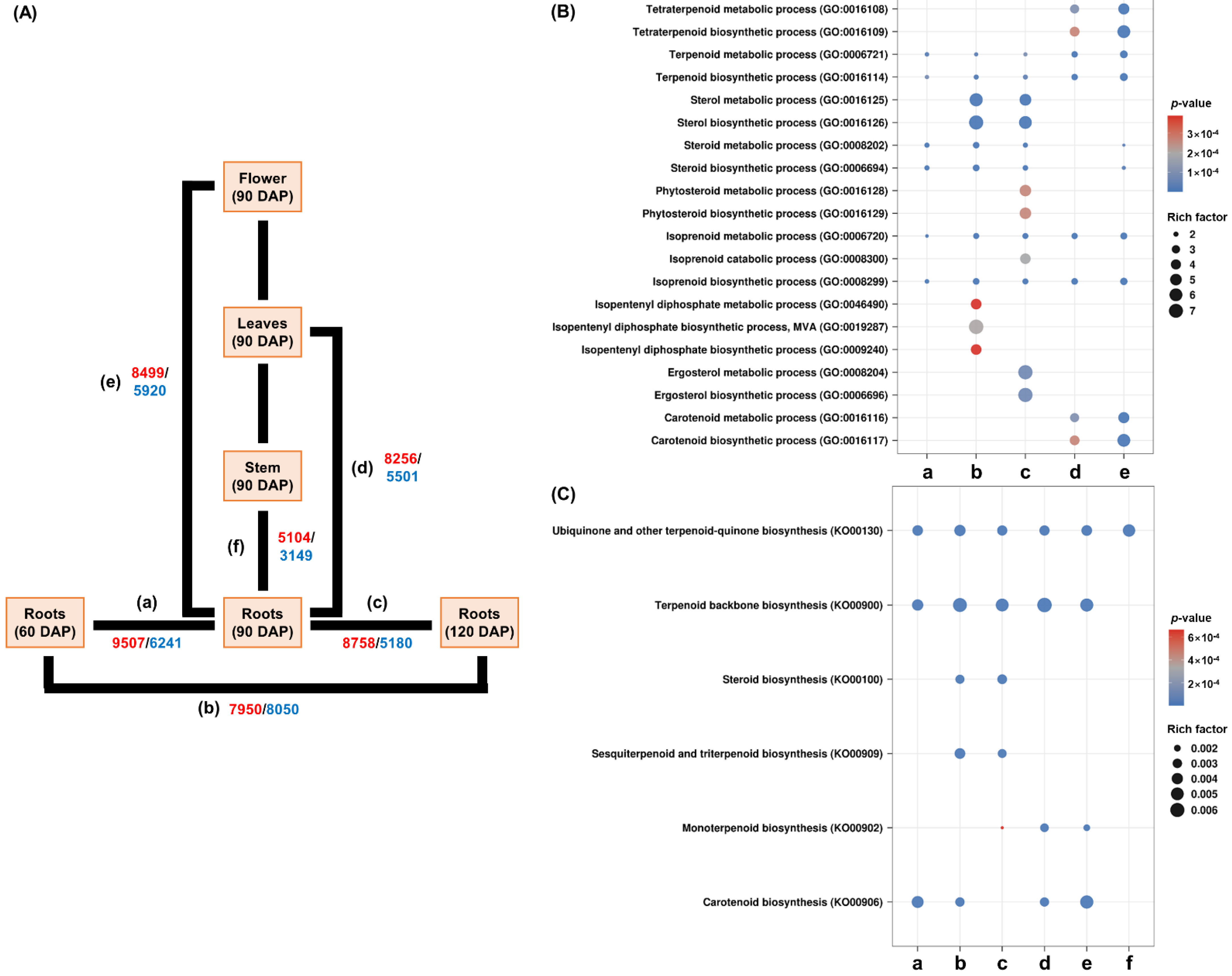
2.2. Analysis of GO and KEGG Terms in Differentially Expressed Genes (DEGs)
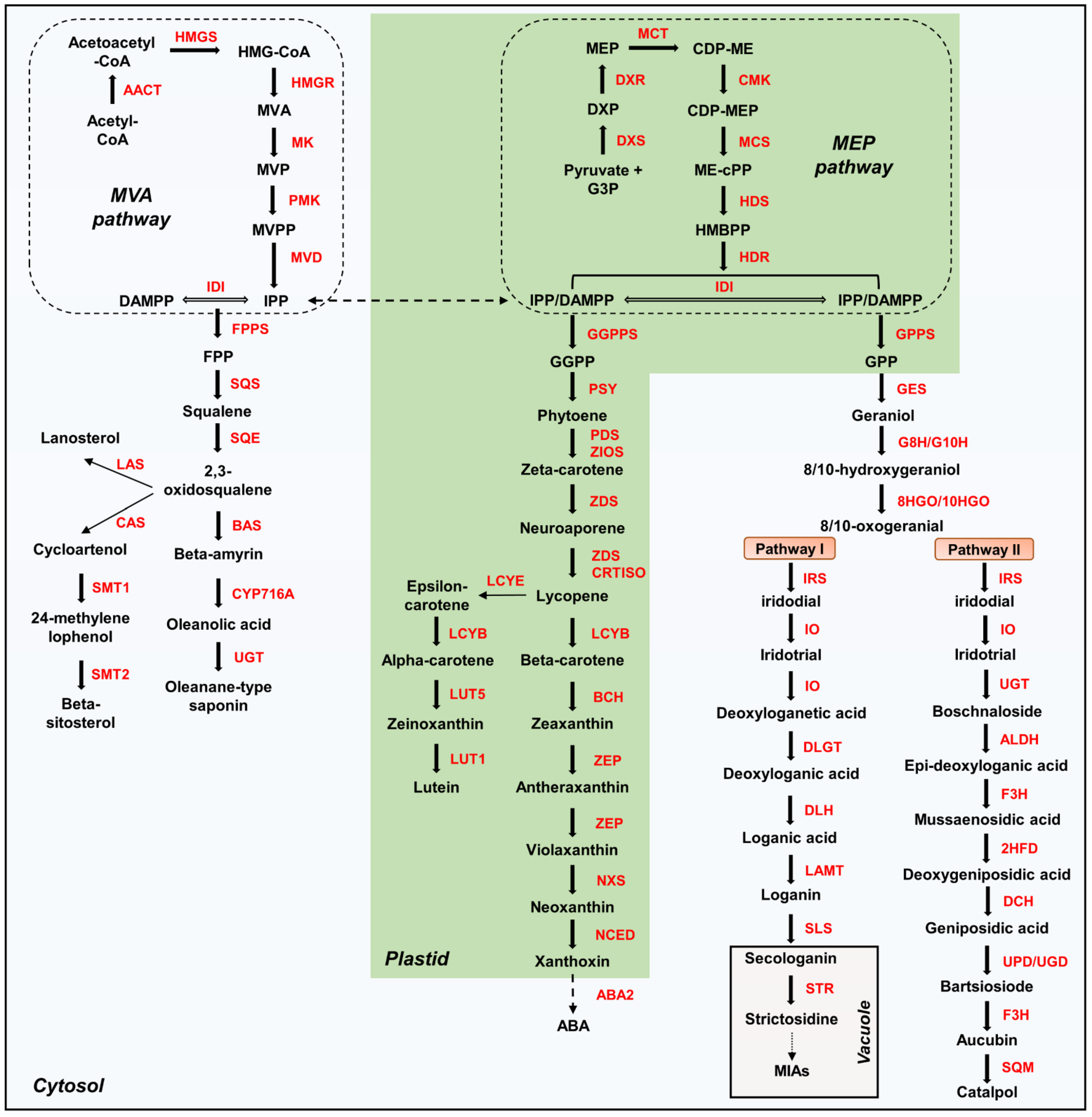
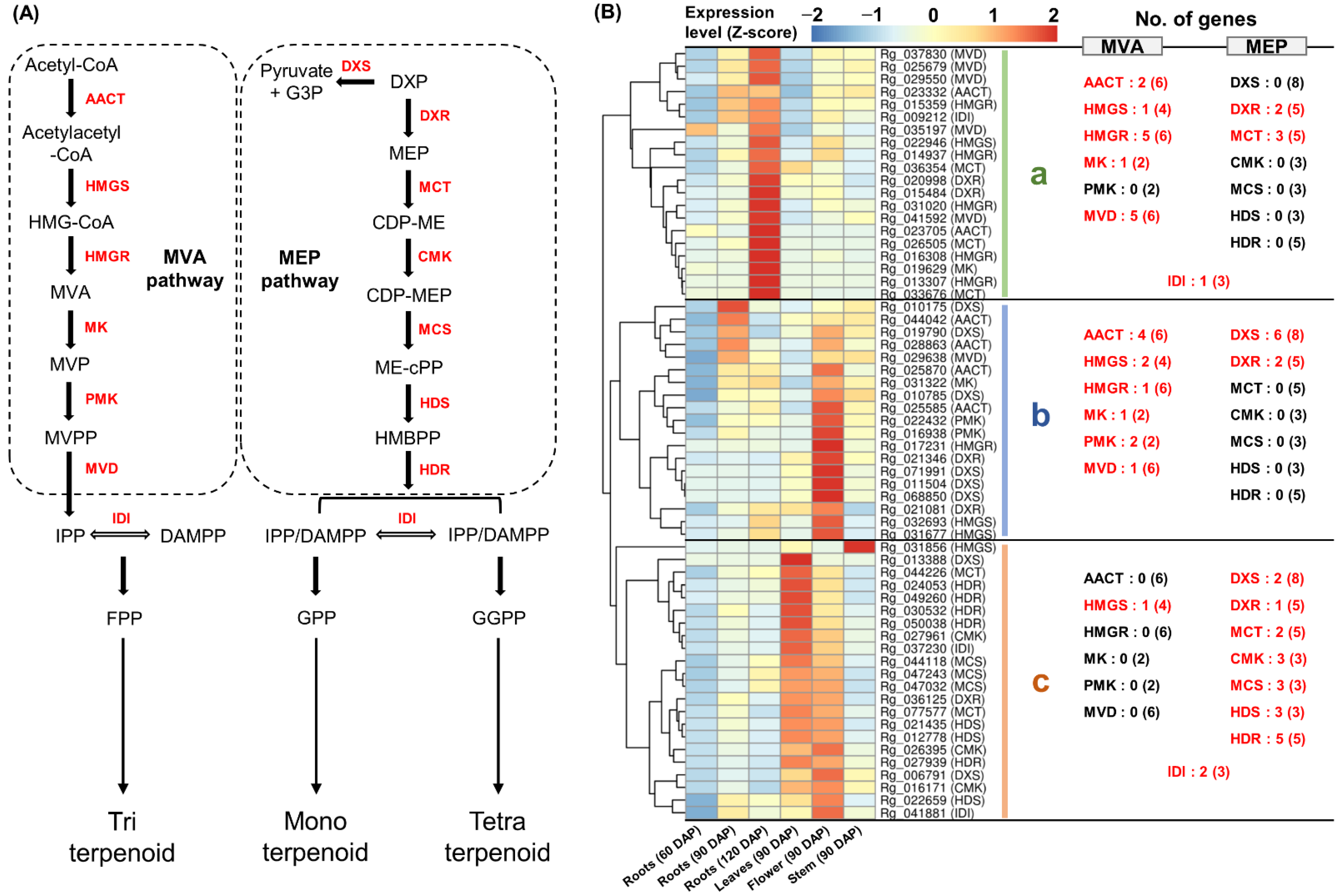
2.3. Expression Analysis of Genes Involved in MVA and MEP Pathways
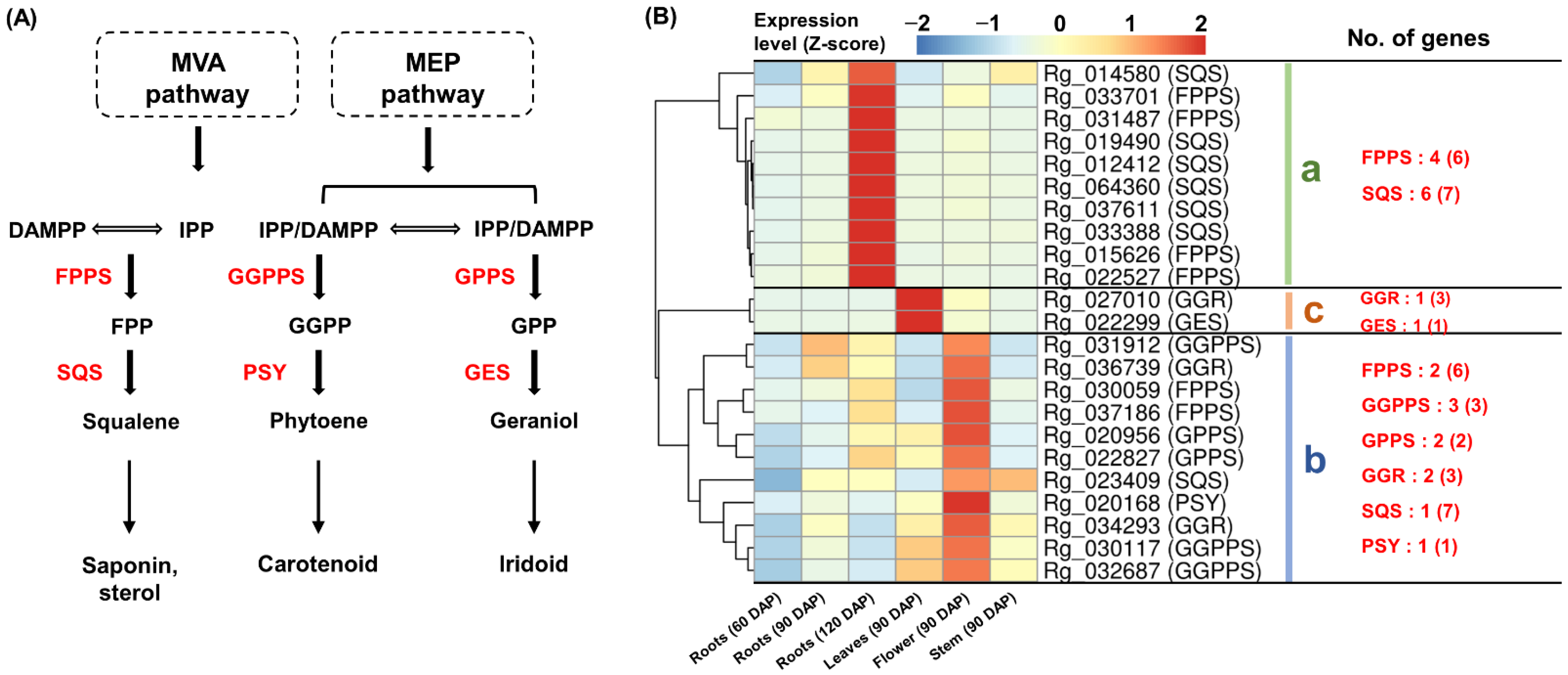
2.4. Specific Co-Expression of TPS-Encoding Genes
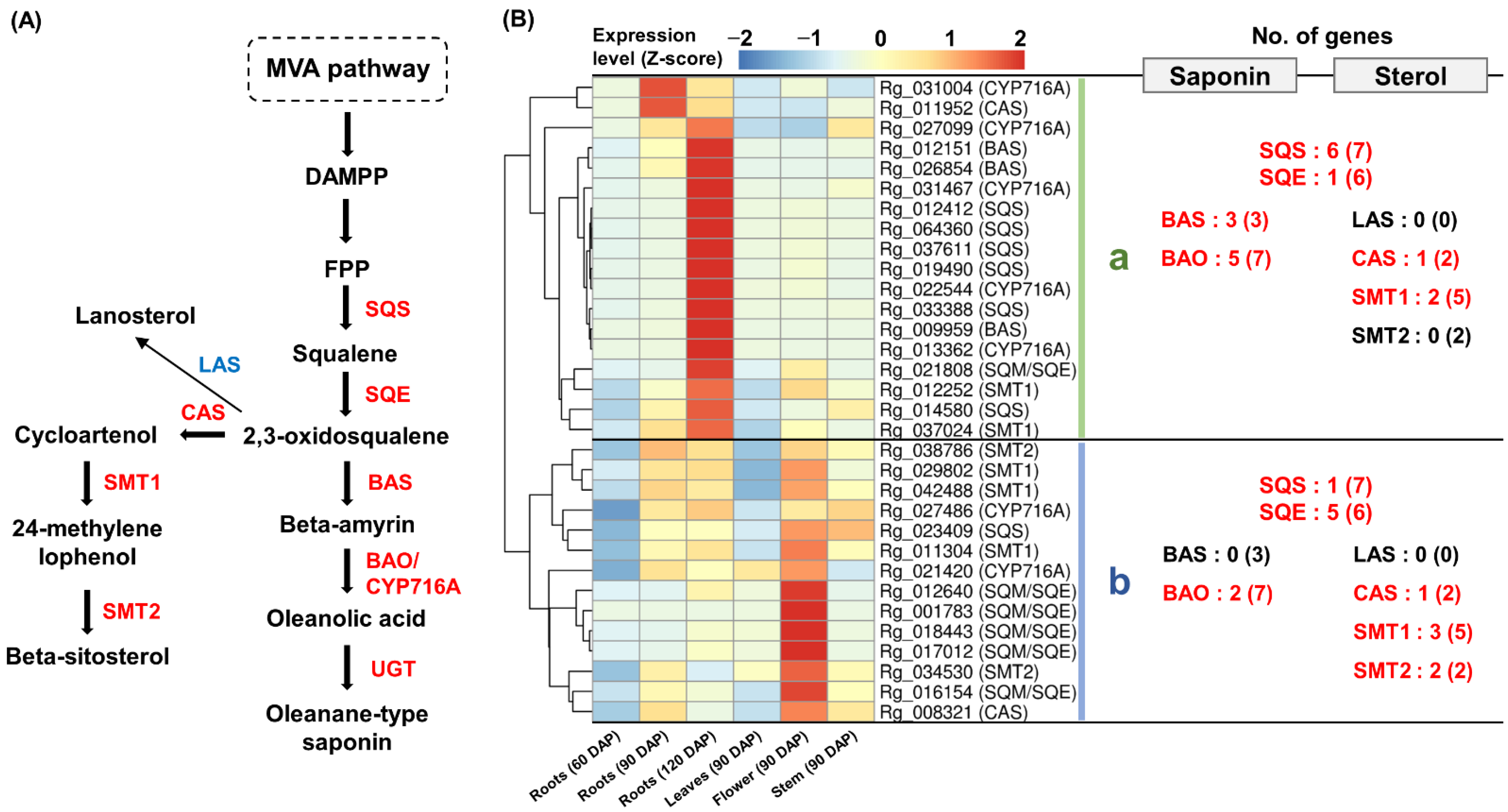
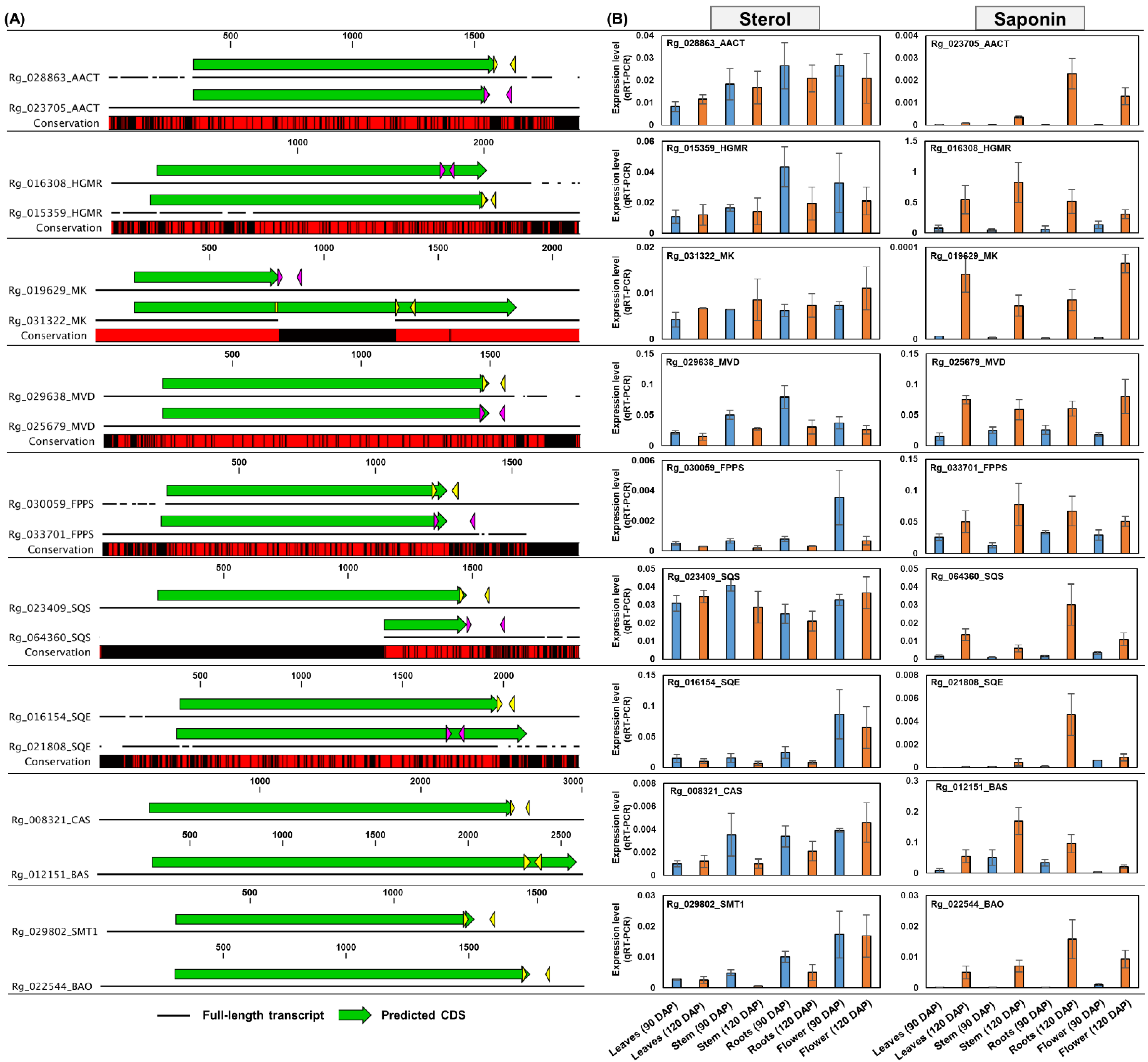
2.5. Expression Analysis of Genes Involved in Sterol and Saponin Biosynthesis Pathways
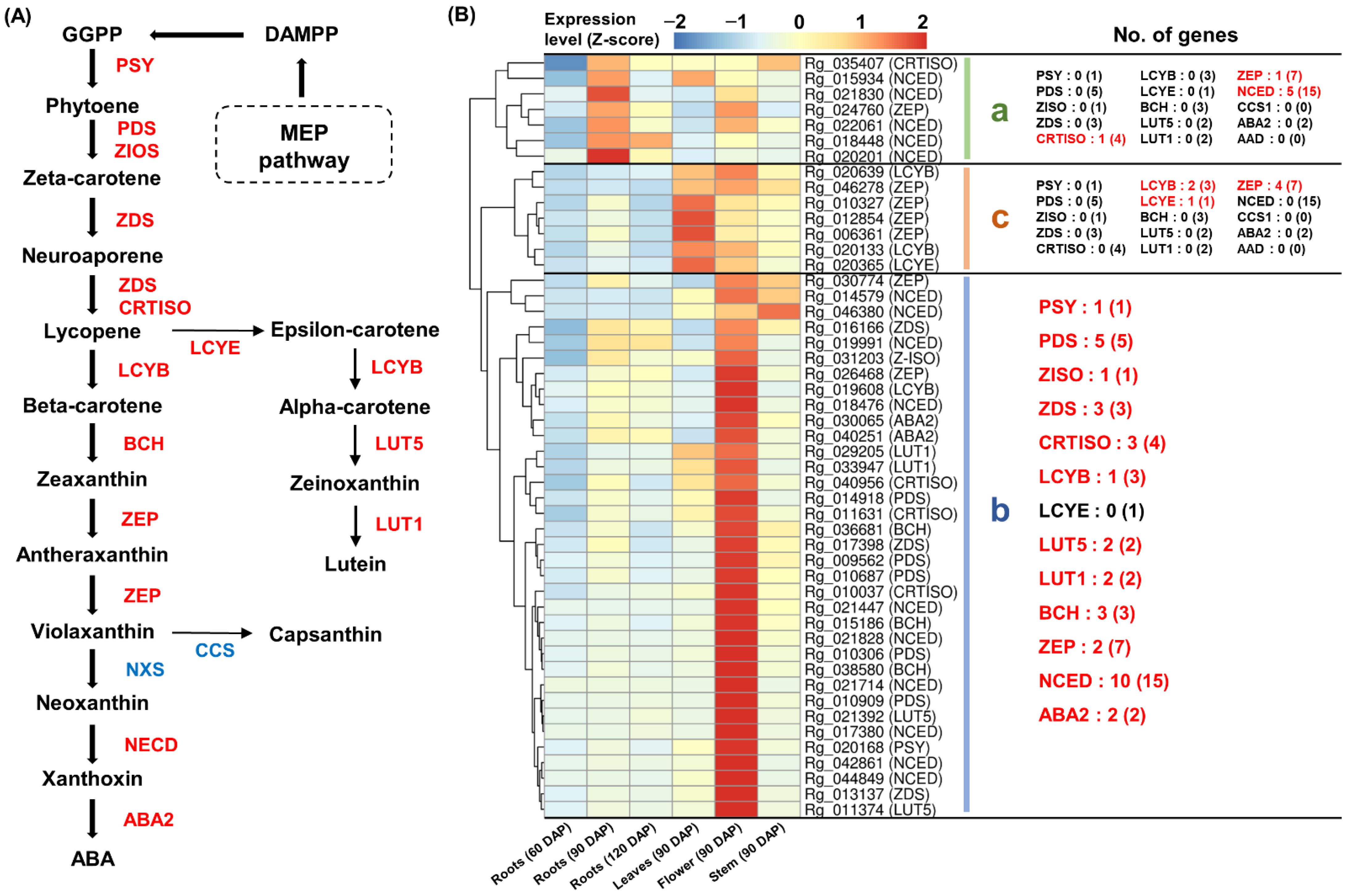
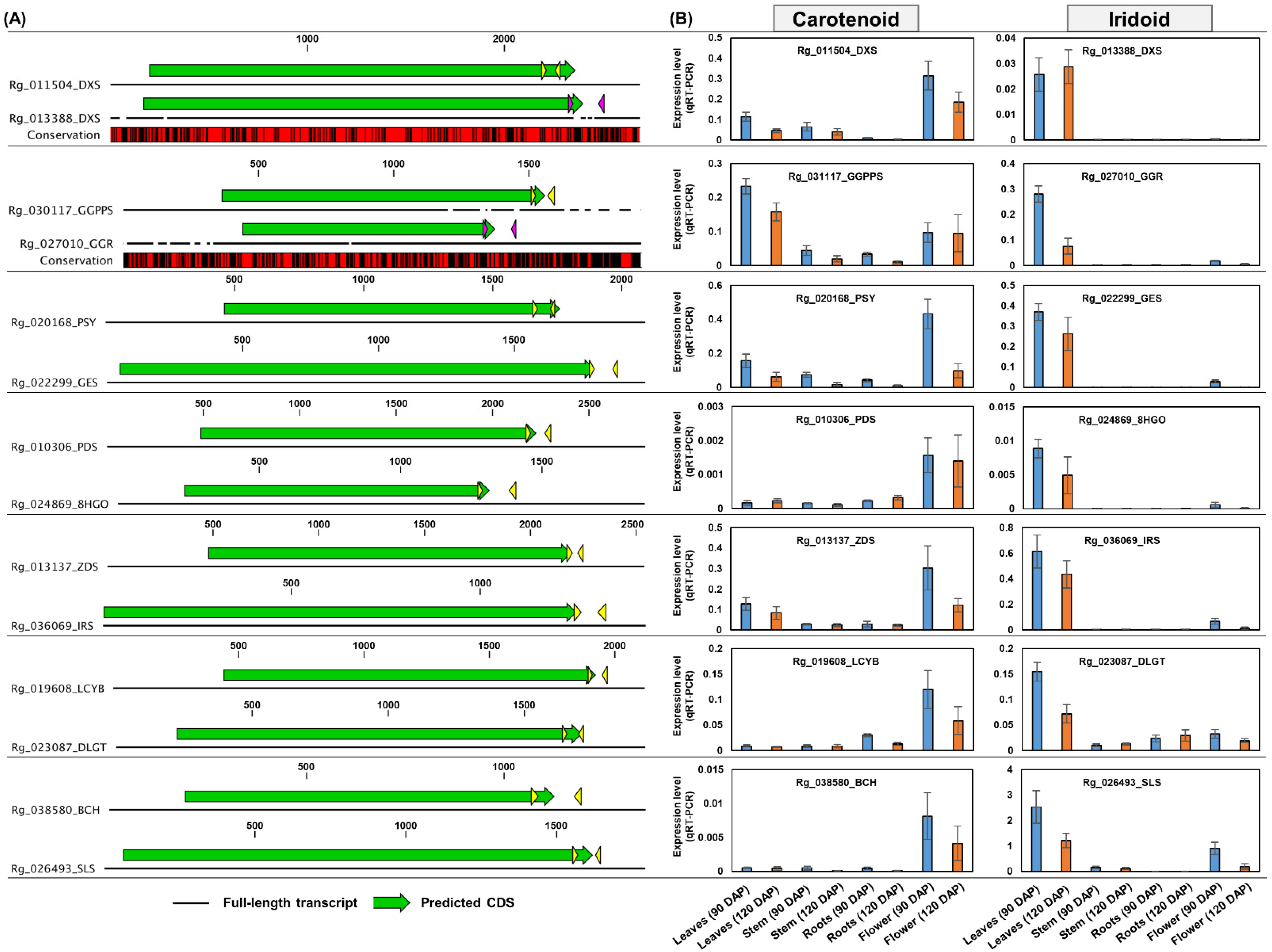
2.6. Expression Analysis of Genes Responsible for the Carotenoid Pathway
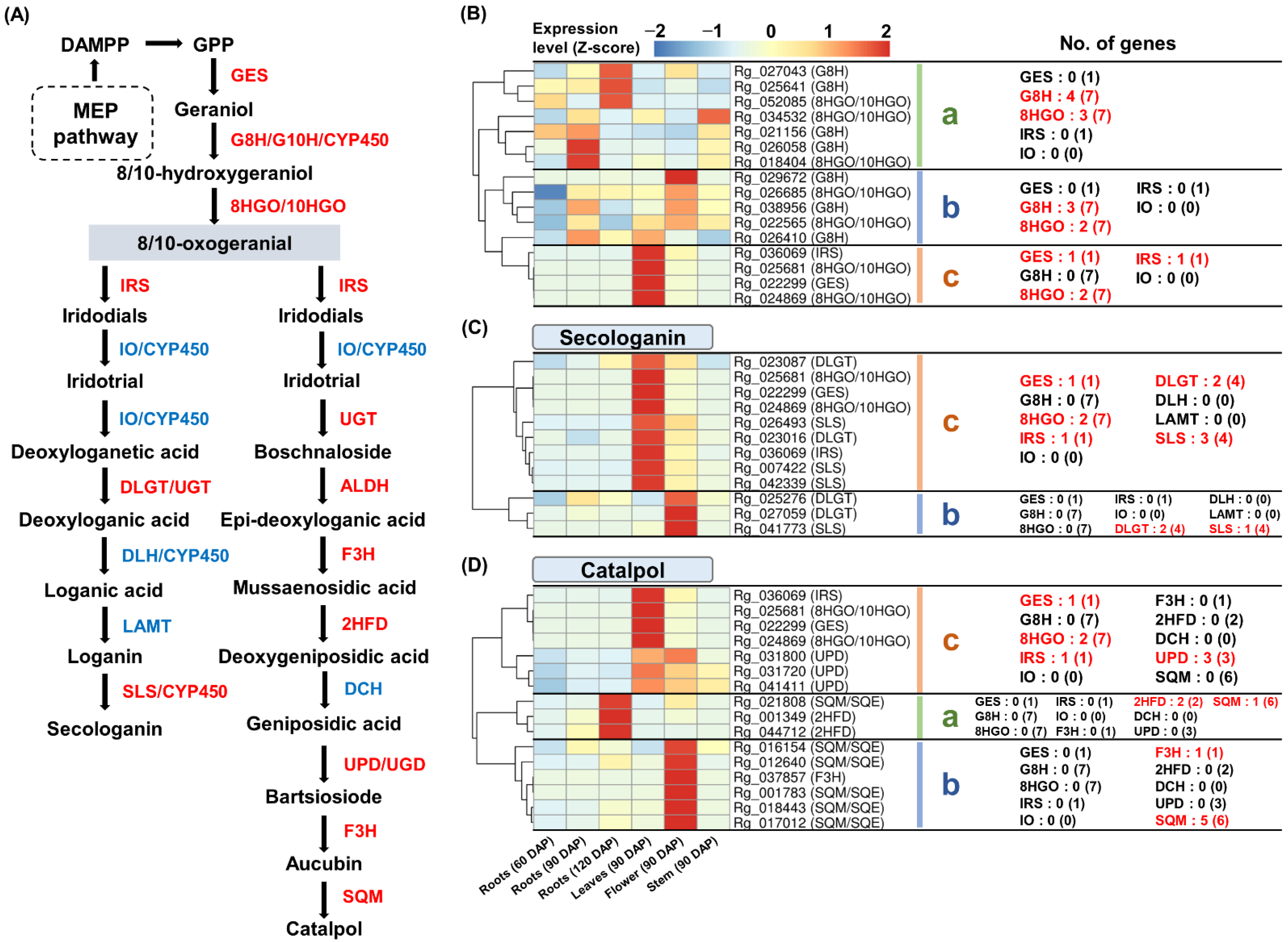
2.7. Identification and Expression Analysis of Iridoid Biosynthesis Genes
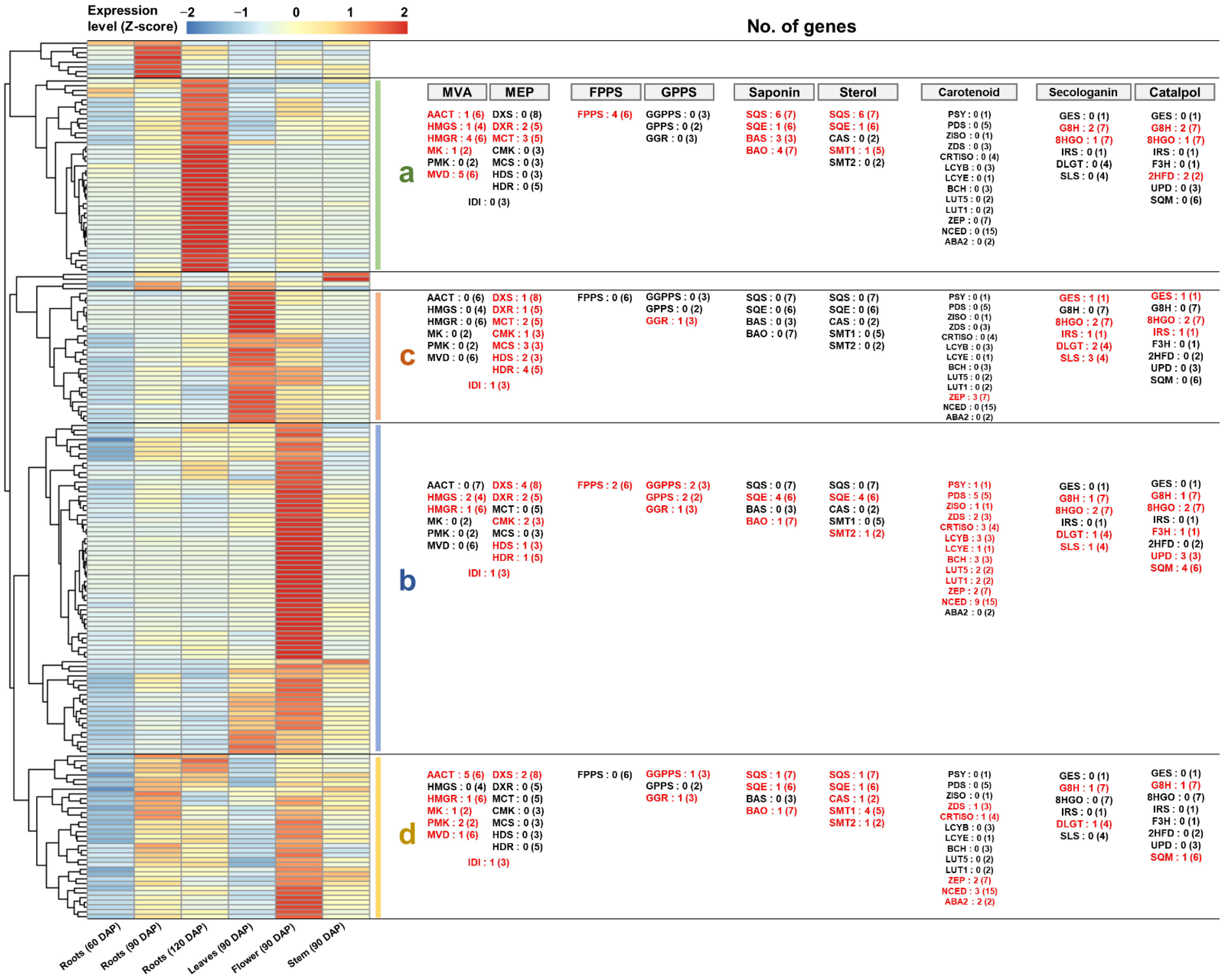
2.8. Comprehensive Co-Expression Analysis of Genes Involved in Terpenoid Biosynthesis
2.9. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Test of Homologous Genes Encoding Major Enzymes in Terpenoid Metabolism
3. Discussion
3.1. Number of Abundant Transcripts in R. glutinosa
3.2. Differential Expression of Homologous Genes for Specific Terpenoid Biosynthesis
3.3. Activity of the Saponin Pathway in Triterpenoid Metabolism
3.4. Carotenoid Biosynthesis from Tetraterpenoid Metabolism
3.5. Biosynthesis of Iridoids from Monoterpenoid Metabolism
4. Conclusions
5. Materials and Methods
5.1. Plant Materials and RNA Preparation
5.2. Production of Transcriptome Data
5.3. Construction of Unigene Set
5.4. Expression Profiling and DEG Analysis
5.5. GO and KEGG Enrichment Analysis
5.6. Analysis of qRT-PCR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| IPP | Isopentenyl pyrophosphate |
| DAMPP | Dimethylallyl pyrophosphate |
| MVA | Mevalonate phosphate |
| MEP | Methylerythritol 4-phosphate |
| TPS | Terpene synthase |
| PT | Prenyltransferase |
| TPC | Terpene cyclase |
| RNA-Seq | RNA sequencing |
| CCS | Circular consensus subread |
| HQ | High quality |
| CDS | Coding region |
| GO | Gene Ontology |
| TPM | Transcripts per million |
| DAP | Day after planting |
| DEGs | Differentially expressed genes |
References
- Lu, X.; Tang, K.; Li, P. Plant metabolic engineering strategies for the production of pharmaceutical terpenoids. Front. Plant Sci. 2016, 7, 1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vranová, E.; Coman, D.; Gruissem, W. Structure and dynamics of the isoprenoid pathway network. Mol. Plant 2012, 5, 318–333. [Google Scholar] [CrossRef] [Green Version]
- Ludwiczuk, A.; Skalicka-Woźniak, K.; Georgiev, M. Terpenoids. In Pharmacognosy; Elsevier: Amsterdam, The Netherlands, 2017; pp. 233–266. [Google Scholar] [CrossRef]
- Moses, T.; Pollier, J.; Thevelein, J.M.; Goossens, A. Bioengineering of plant (tri) terpenoids: From metabolic engineering of plants to synthetic biology in vivo and in vitro. New Phytol. 2013, 200, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.-C.; Kuo, Y.-H.; Jan, J.-T.; Liang, P.-H.; Wang, S.-Y.; Liu, H.-G.; Lee, C.-K.; Chang, S.-T.; Kuo, C.-J.; Lee, S.-S. Specific plant terpenoids and lignoids possess potent antiviral activities against severe acute respiratory syndrome coronavirus. J. Med. Chem. 2007, 50, 4087–4095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karunanithi, P.S.; Zerbe, P. Terpene synthases as metabolic gatekeepers in the evolution of plant terpenoid chemical diversity. Front. Plant Sci. 2019, 10, 1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Wei, X.; Qi, Q.; Jia, W.; Zhao, M.; Wang, H.; Zhou, Y.; Duan, H. Study of Terpenoid Synthesis and Prenyltransferase in Roots of Rehmannia glutinosa Based on iTRAQ Quantitative Proteomics. Front. Plant Sci. 2021, 12, 1185. [Google Scholar] [CrossRef]
- Phillips, M.A.; D’Auria, J.C.; Gershenzon, J.; Pichersky, E. The Arabidopsis thaliana type I isopentenyl diphosphate isomerases are targeted to multiple subcellular compartments and have overlapping functions in isoprenoid biosynthesis. Plant Cell 2008, 20, 677–696. [Google Scholar] [CrossRef] [Green Version]
- Bohlmann, J.; Meyer-Gauen, G.; Croteau, R. Plant terpenoid synthases: Molecular biology and phylogenetic analysis. Proc. Natl. Acad. Sci. USA 1998, 95, 4126–4133. [Google Scholar] [CrossRef] [Green Version]
- Christianson, D.W. Structural and chemical biology of terpenoid cyclases. Chem. Rev. 2017, 117, 11570–11648. [Google Scholar] [CrossRef] [Green Version]
- Kopcsayová, D.; Vranová, E. Functional gene network of prenyltransferases in Arabidopsis thaliana. Molecules 2019, 24, 4556. [Google Scholar] [CrossRef] [Green Version]
- Iijima, Y.; Gang, D.R.; Fridman, E.; Lewinsohn, E.; Pichersky, E. Characterization of geraniol synthase from the peltate glands of sweet basil. Plant Physiol. 2004, 134, 370–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Li, C.; Wang, L.; Fei, Y.; Qin, W. Analysis of Centranthera grandiflora benth transcriptome explores genes of catalpol, acteoside and azafrin biosynthesis. Int. J. Mol. Sci. 2019, 20, 6034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nisar, N.; Li, L.; Lu, S.; Khin, N.C.; Pogson, B.J. Carotenoid metabolism in plants. Mol. Plant 2015, 8, 68–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shitiz, K.; Sharma, N.; Pal, T.; Sood, H.; Chauhan, R.S. NGS transcriptomes and enzyme inhibitors unravel complexity of picrosides biosynthesis in Picrorhiza kurroa Royle ex. Benth. PLoS ONE 2015, 10, e0144546. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, K.; Dong, L.; Navrot, N.; Schneider, T.; Burlat, V.; Pollier, J.; Woittiez, L.; Van Der Krol, S.; Lugan, R.; Ilc, T. The seco-iridoid pathway from Catharanthus roseus. Nat. Commun. 2014, 5, 3606. [Google Scholar] [CrossRef] [Green Version]
- Group, A.P.; Chase, M.W.; Christenhusz, M.J.; Fay, M.F.; Byng, J.; Judd, W.; Soltis, D.; Mabberley, D.; Sennikov, A.; Soltis, P. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. 2016, 181, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.-X.; Li, M.-X.; Jia, Z.-P. Rehmannia glutinosa: Review of botany, chemistry and pharmacology. J. Ethnopharmacol. 2008, 117, 199–214. [Google Scholar] [CrossRef]
- Sun, P.; Song, S.; Zhou, L.; Zhang, B.; Qi, J.; Li, X. Transcriptome analysis reveals putative genes involved in iridoid biosynthesis in Rehmannia glutinosa. Int. J. Mol. Sci. 2012, 13, 13748–13763. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Kim, D.; Park, C.; Han, S.; Park, C.; Park, H.; Kim, Y.; Park, C. A disease resistance and high yielding Rehmannia glutinosa Libosch. ex Steud. cultivar ‘Tokang’. Korean J. Breed. Sci. 2017, 49, 36–40. [Google Scholar] [CrossRef]
- Hoang, N.V.; Furtado, A.; Mason, P.J.; Marquardt, A.; Kasirajan, L.; Thirugnanasambandam, P.P.; Botha, F.C.; Henry, R.J. A survey of the complex transcriptome from the highly polyploid sugarcane genome using full-length isoform sequencing and de novo assembly from short read sequencing. BMC Genom. 2017, 18, 395. [Google Scholar] [CrossRef]
- Li, J.; Harata-Lee, Y.; Denton, M.D.; Feng, Q.; Rathjen, J.R.; Qu, Z.; Adelson, D.L. Long read reference genome-free reconstruction of a full-length transcriptome from Astragalus membranaceus reveals transcript variants involved in bioactive compound biosynthesis. Cell Discov. 2017, 3, 17031. [Google Scholar] [CrossRef] [PubMed]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Prjibelski, A.D. rnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. Gigascience 2019, 8, giz100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Wang, Y.; Guo, F.; Zhan, L.; Mohr, T.; Cheng, P.; Huo, N.; Gu, R.; Pei, D.; Sun, J. Deep sequencing and transcriptome analyses to identify genes involved in secoiridoid biosynthesis in the Tibetan medicinal plant Swertia mussotii. Sci. Rep. 2017, 7, 43108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Dong, C.; Song, C.; Wang, X.; Zheng, X.; Niu, Y.; Chen, S.; Feng, W. De novo genome assembly of the potent medicinal plant Rehmannia glutinosa using nanopore technology. Comput. Struct. Biotechnol. J. 2021, 19, 3954–3963. [Google Scholar] [CrossRef] [PubMed]
- Vranová, E.; Coman, D.; Gruissem, W. Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu. Rev. Plant Biol. 2013, 64, 665–700. [Google Scholar] [CrossRef]
- Opitz, S.; Nes, W.D.; Gershenzon, J. Both methylerythritol phosphate and mevalonate pathways contribute to biosynthesis of each of the major isoprenoid classes in young cotton seedlings. Phytochemistry 2014, 98, 110–119. [Google Scholar] [CrossRef]
- Jin, H.; Song, Z.; Nikolau, B.J. Reverse genetic characterization of two paralogous acetoacetyl CoA thiolase genes in Arabidopsis reveals their importance in plant growth and development. Plant J. 2012, 70, 1015–1032. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Lee, O.R.; Oh, J.Y.; Jang, M.-G.; Yang, D.-C. Functional analysis of 3-hydroxy-3-methylglutaryl coenzyme a reductase encoding genes in triterpene saponin-producing ginseng. Plant Physiol. 2014, 165, 373–387. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.; Ward, B.L.; Bostock, R.M. Differential induction and suppression of potato 3-hydroxy-3-methylglutaryl coenzyme A reductase genes in response to Phytophthora infestans and to its elicitor arachidonic acid. Plant Cell 1992, 4, 1333–1344. [Google Scholar] [CrossRef] [Green Version]
- Cunillera, N.; Arró, M.; Delourme, D.; Karst, F.; Boronat, A.; Ferrer, A. Arabidopsis thaliana Contains Two Differentially Expressed Farnesyl-Diphosphate Synthase Genes (∗). J. Biol. Chem. 1996, 271, 7774–7780. [Google Scholar] [CrossRef] [Green Version]
- Closa, M.; Vranová, E.; Bortolotti, C.; Bigler, L.; Arró, M.; Ferrer, A.; Gruissem, W. The Arabidopsis thaliana FPP synthase isozymes have overlapping and specific functions in isoprenoid biosynthesis, and complete loss of FPP synthase activity causes early developmental arrest. Plant J. 2010, 63, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-D.; Han, J.-Y.; Huh, G.H.; Choi, Y.-E. Expression and functional characterization of three squalene synthase genes associated with saponin biosynthesis in Panax ginseng. Plant Cell Physiol. 2011, 52, 125–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.-Y.; In, J.-G.; Kwon, Y.-S.; Choi, Y.-E. Regulation of ginsenoside and phytosterol biosynthesis by RNA interferences of squalene epoxidase gene in Panax ginseng. Phytochemistry 2010, 71, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.R.; Shilpashree, H.; Nagegowda, D.A. Terpene moiety enhancement by overexpression of geranyl (geranyl) diphosphate synthase and geraniol synthase elevates monomeric and dimeric monoterpene indole alkaloids in transgenic Catharanthus roseus. Front. Plant Sci. 2018, 9, 942. [Google Scholar] [CrossRef]
- Zhang, F.; Liu, W.; Xia, J.; Zeng, J.; Xiang, L.; Zhu, S.; Zheng, Q.; Xie, H.; Yang, C.; Chen, M. Molecular characterization of the 1-deoxy-D-xylulose 5-phosphate synthase gene family in Artemisia annua. Front. Plant Sci. 2018, 9, 952. [Google Scholar] [CrossRef]
- Okada, A.; Shimizu, T.; Okada, K.; Kuzuyama, T.; Koga, J.; Shibuya, N.; Nojiri, H.; Yamane, H. Elicitor induced activation of the methylerythritol phosphate pathway toward phytoalexins biosynthesis in rice. Plant Mol. Biol. 2007, 65, 177–187. [Google Scholar] [CrossRef]
- You, M.; Lee, Y.; Kim, J.; Baek, S.; Jeon, Y.; Lim, S.; Ha, S. The organ-specific differential roles of rice DXS and DXR, the first two enzymes of the MEP pathway, in carotenoid metabolism in Oryza sativa leaves and seeds. BMC Plant Biol. 2020, 20, 167. [Google Scholar] [CrossRef] [Green Version]
- Lois, L.M.; Rodríguez-Concepción, M.; Gallego, F.; Campos, N.; Boronat, A. Carotenoid biosynthesis during tomato fruit development: Regulatory role of 1-deoxy-D-xylulose 5-phosphate synthase. Plant J. 2000, 22, 503–513. [Google Scholar] [CrossRef]
- Rai, A.; Smita, S.S.; Singh, A.K.; Shanker, K.; Nagegowda, D.A. Heteromeric and homomeric geranyl diphosphate synthases from Catharanthus roseus and their role in monoterpene indole alkaloid biosynthesis. Mol. Plant 2013, 6, 1531–1549. [Google Scholar] [CrossRef] [Green Version]
- Estévez, J.M.; Cantero, A.; Reindl, A.; Reichler, S.; León, P. 1-Deoxy-D-xylulose-5-phosphate synthase, a limiting enzyme for plastidic isoprenoid biosynthesis in plants. J. Biol. Chem. 2001, 276, 22901–22909. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, A.; Shimada, H.; Masuda, T.; Ohta, H.; Takamiya, K.-I. Two distinct isopentenyl diphosphate isomerases in cytosol and plastid are differentially induced by environmental stresses in tobacco. FEBS Lett. 2001, 506, 61–64. [Google Scholar] [CrossRef] [Green Version]
- Iñiguez, L.P.; Hernández, G. The evolutionary relationship between alternative splicing and gene duplication. Front. Genet. 2017, 8, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alex, D.; Bach, T.J.; Chye, M.L. Expression of Brassica juncea 3-hydroxy-3-methylglutaryl CoA synthase is developmentally regulated and stress-responsive. Plant J. 2000, 22, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, K.; Melton, R.; Leggett, M.; Daniels, M.; Osbourn, A. Compromised disease resistance in saponin-deficient plants. Proc. Natl. Acad. Sci. USA 1999, 96, 12923–12928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asada, K.; Salim, V.; Masada-Atsumi, S.; Edmunds, E.; Nagatoshi, M.; Terasaka, K.; Mizukami, H.; De Luca, V. A 7-deoxyloganetic acid glucosyltransferase contributes a key step in secologanin biosynthesis in Madagascar periwinkle. Plant Cell 2013, 25, 4123–4134. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Dai, X.; Su, S.; Yan, H.; Guo, S.; Qian, D.; Duan, J.A. Investigation of dynamic accumulation and regularity of nine glycosides and saccharides in Rehmannia glutinosa by rapid quantitative analysis technology. J. Sep. Sci. 2019, 42, 1489–1499. [Google Scholar] [CrossRef]
- Wang, Y.; Liao, D.; Qin, M.; Li, X.E. Simultaneous determination of catalpol, aucubin, and geniposidic acid in different developmental stages of Rehmannia glutinosa leaves by high performance liquid chromatography. J. Anal. Methods Chem. 2016, 2016, 4956589. [Google Scholar] [CrossRef] [Green Version]
- Vennapusa, A.R.; Somayanda, I.M.; Doherty, C.J.; Krishna Jagadish, S. A universal method for high-quality RNA extraction from plant tissues rich in starch, proteins and fiber. Sci. Rep. 2020, 10, 16887. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Lawrence Berkeley National Lab. (LBNL): Berkeley, CA, USA, 2014. [Google Scholar]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Net. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, 116–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, 182–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Cheadle, C.; Vawter, M.P.; Freed, W.J.; Becker, K.G. Analysis of microarray data using Z score transformation. J. Mol. Diagn. 2003, 5, 73–81. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Unigenes | Predicted CDS | Deduced Protein | |
|---|---|---|---|
| Unigene No. | 140,335 | 58,949 | 58,946 |
| Total length (bp) | 149,495,081 | 57,594,645 | 19,198,215 |
| Minimum length (bp) | 165 | 255 | 85 |
| Maximum length (bp) | 16,911 | 16,239 | 5413 |
| Average length (bp) | 1065 | 977 | 326 |
| N50 (bp) | 1821 | 1287 | 429 |
| GC Ratio (%) | 39.72 | 44.14 | 152 |
| BUSCO ver5 | 98% | - | - |
| Database | Program and Parameters | Number of Annotated Genes | Percentage of Total Genes |
|---|---|---|---|
| NCBI nr proteins | DIAMOND, cutoff e-value 1E-5 | 77,747 | 55.40% |
| GO | Blast2GO, default parameters | 51,296 | 36.55% |
| InterProScan | InterProScan | 45,663 | 32.54% |
| KEGG | KAAS, SBH method | 30,226 | 21.54% |
| Araport11 | BLASTX, cutoff e-value 1E-5 | 61,242 | 43.64% |
| Total | 78,559 | 55.98% | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, J.-N.; Han, J.-W.; Yang, S.-H.; Lee, S.-M. Co-Expression Analysis Reveals Differential Expression of Homologous Genes Associated with Specific Terpenoid Biosynthesis in Rehmannia glutinosa. Genes 2022, 13, 1092. https://doi.org/10.3390/genes13061092
Kang J-N, Han J-W, Yang S-H, Lee S-M. Co-Expression Analysis Reveals Differential Expression of Homologous Genes Associated with Specific Terpenoid Biosynthesis in Rehmannia glutinosa. Genes. 2022; 13(6):1092. https://doi.org/10.3390/genes13061092
Chicago/Turabian StyleKang, Ji-Nam, Jong-Won Han, So-Hee Yang, and Si-Myung Lee. 2022. "Co-Expression Analysis Reveals Differential Expression of Homologous Genes Associated with Specific Terpenoid Biosynthesis in Rehmannia glutinosa" Genes 13, no. 6: 1092. https://doi.org/10.3390/genes13061092