
paPAML: An Improved Computational Tool to Explore Selection Pressure on Protein-Coding Sequences

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. CDS-Extractor and Alignment
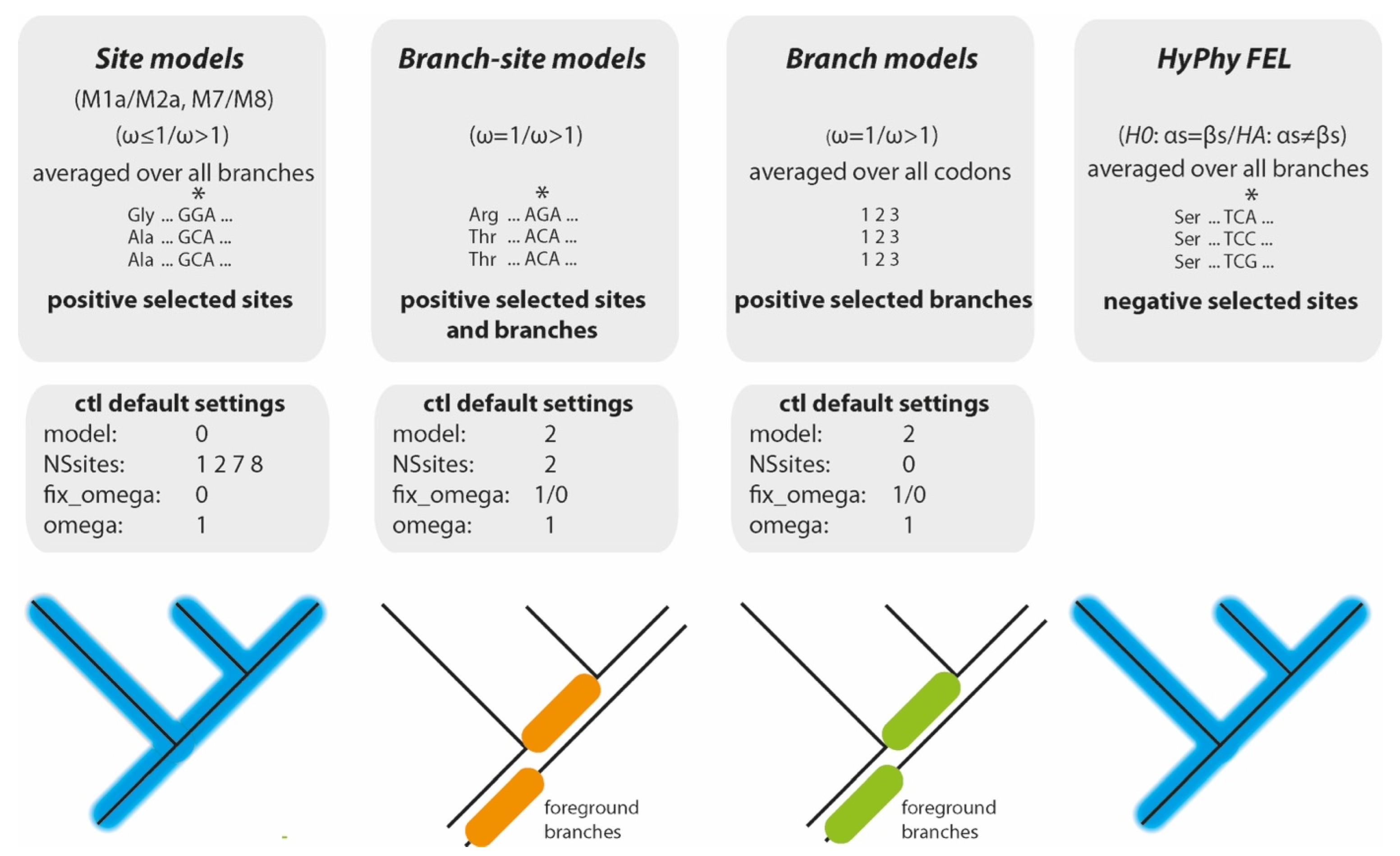
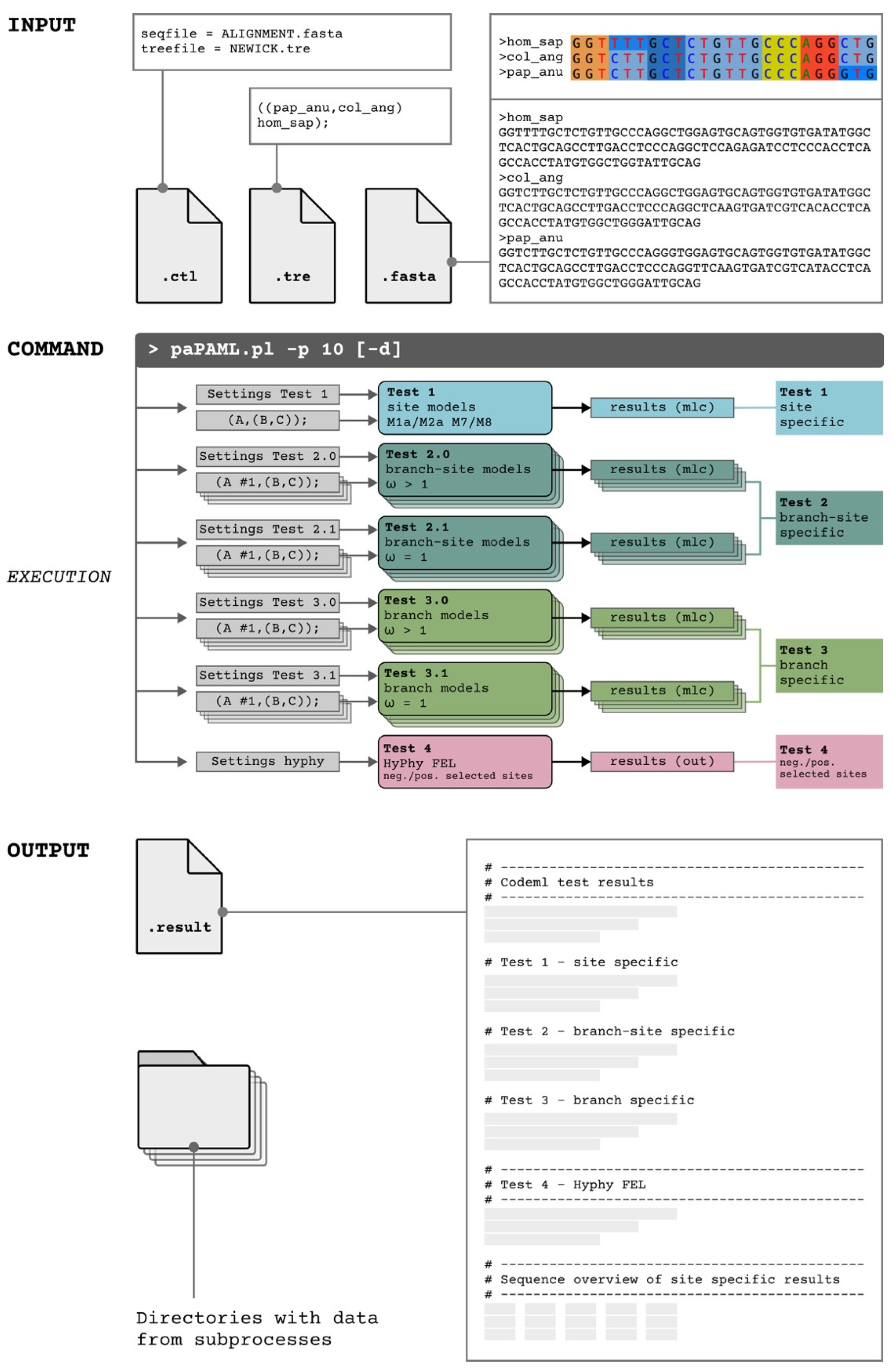
2.2. paPAML
2.3. Data Requirements (paPAML)
2.4. Necessary Resources (paPAML)
3. Results and Discussion
3.1. Four Examples of Test Data
3.2. Comparison to Other Selection Analysis Tools
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed] [Green Version]
- Castro-Chavez, F. The rules of variation: Amino acid exchange according to the rotating circular genetic code. J. Theor. Biol. 2010, 264, 711–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strachan, T.; Read, A.P. Human Molecular Genetics, 2nd ed.; Wiley: New York, NY, USA, 1999; p. 576. [Google Scholar]
- Nielsen, R.; Yang, Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 1998, 148, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R.; Goldman, N.; Pedersen, A.M. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 2000, 155, 431–449. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Wong, W.S.W.; Nielsen, R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Nielsen, R.; Yang, Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 2005, 22, 2472–2479. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 1998, 15, 568–573. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R. Synonymous and nonsynonymous rate variation in nuclear genes of mammals. J. Mol. Evol. 1998, 46, 409–418. [Google Scholar] [CrossRef]
- Pond, S.L.K.; Frost, S.D.W.; Muse, S.V. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [Green Version]
- Pond, S.L.K.; Frost, S.D.W. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartig, G.; Churakov, G.; Warren, W.; Brosius, J.; Makałowski, W.; Schmitz, J. Retrophylogenomics place tarsiers on the evolutionary branch of anthropoids. Sci. Rep. 2013, 3, 1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrader, L.; Schmitz, J. The impact of transposable elements in adaptive evolution. Mol. Ecol. 2019, 28, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Raabe, C.A.; Cardoso-Moreira, M.; Brosius, J.; Kaessmann, H.; Schmitz, J. ExoPLOT: Advance in differential expression of alternative human exons. Genomics, 2022; under review. [Google Scholar]
- Pinheiro, D.; Mawhin, M.A.; Prendecki, M.; Woollard, K.J. In-silico analysis of myeloid cells across the animal kingdom reveals neutrophil evolution by colony-stimulating factors. eLife 2020, 9, 60214. [Google Scholar] [CrossRef] [PubMed]
- Massingham, T.; Goldman, N. Detecting amino acid sites under positive selection and purifying selection. Genetics 2005, 169, 1753–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, D.; Schmitz, J.; Fischer, K.; Granado, D.; Groh, A.C.; Krausel, V.; Lüttgenau, S.M.; Amelung, T.M.; Pavenstädt, H.; Weide, T. Evolution of renal-disease factor APOL1 results in cis and trans orientations at the endoplasmic reticulum that both show cytotoxic effects. Mol. Biol. Evol. 2021, 38, 4962–4976. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE v5 enables improved estimates of phylogenetic tree confidence by ensemble bootstrapping. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lee, Y.H.; Ota, T.; Vacquier, V.D. Positive selection is a general phenomenon in the evolution of abalone sperm lysin. Mol. Biol. Evol. 1995, 12, 231–238. [Google Scholar]
- Gao, F.; Chen, C.; Arab, D.A.; Du, Z.; He, Y.; Ho, S.Y.W. EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evol. 2019, 9, 3891–3898. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, E.; Almeida, D.; Escalona, T.; Khan, I.; Vasconcelos, V.; Antunes, A. LMAP: Lightweight multigene analyses in PAML. BMC Bioinform. 2016, 17, 354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schott, R.K.; Gow, D.; Chang, B.S.W. BlastPhyMe: A toolkit for rapid generation and analysis of protein-coding sequence datasets. bioRxiv 2016. [Google Scholar] [CrossRef] [Green Version]
- Dyachkova, M.S.; Chekalin, E.V.; Danilenko, V.N. Positive selection in Bifidobacterium genes drives specis-specific host-bacteria communication. Front. Microbiol. 2019, 10, 2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| paPAML-VERSION 1.23 | |
|---|---|
| paPAML.pl -p threads | [-f controlfiles] [-t tests] [-s significance] [-d] {codemlparams} |
| paPAML.pl -i | [provides status information of runs in a new terminal window] |
| paPAML.pl -c | [removes all temporal folders] |
| CTRL-c | [interrupts the run and all subprocesses] |
| where: | |
| runs | The number of parallel runs. |
| controlfiles | A list of control files. It is assumed they are named with the suffix “.ctl”. If not given, all files with that suffix are used for calculation. |
| tests | The used tests (1, 2, 3 or h for HyPhy) to run the data. They can be written like “1” or “12” (order is not important, default: 123h). |
| significance | The maximum p-value to print sites and branches/trees under selection. Also used for printing BEB and HyPhy results (default: 0.05). |
| -d | Original result directories (PAML, HyPhy) are stored. |
| -I | Info about current runs. |
| -c | Cleans all temporary folders. |
| codemlparams | If not provided additional parameters for the ctl file, the default parameters will be used (see below). |
| Example: -Mgene 9 -rho 34 | |
| default codeml-parameters | |
| -CodonFreq | 2 |
| -Malpha | 0 |
| -Mgene | 0 |
| -RateAncestor | 0 |
| -Small_Diff | 0.5e-6 |
| -aaDist | 0 |
| -alpha | 0 |
| -cleandata | 1 |
| -clock | 0 |
| -fix_alpha | 1 |
| -fix_blength | −1 |
| -fix_rho | 1 |
| -getSE | 0 |
| -icode | 0 |
| -method | 0 |
| -ndata | 1 |
| -omega | 1 |
| -outfile | mlc |
| -rho | 0 |
| -runmode | 0 |
| -seqtype | 1 |
| Dataset | Number of Sequences | Number of Codons | Overall Runtime paPAML (min) | Longest Runtime Codeml (min) | Overall Runtime Codeml (min) |
|---|---|---|---|---|---|
| GINS3 | 3 | 39 | <1 | <1 | <1 |
| CEBPE | 30 | 281 | 198 | 1205 | 3032 |
| Lysin | 25 | 120 | 115 | 405 | 867 |
| APOL1–4 | 20 | 312 | 79 | 447 | 1143 |
| Dataset | Number of Codons in Sequences | Number of Positively Selected Codons (Site Models) | Number of Positively Selected Codons (Branch-Site Model) | Number of Positively Selected Codons (HyPhy FEL) | Number of Negatively Selected Codons (HyPhy FEL) | Number of Branches under Positive Selection |
|---|---|---|---|---|---|---|
| GINS3 | 39 | 8 | 0 | 0 | 0 | 2 (branch-site model) |
| CEBPE | 281 | 0 | 0 | 0 | 68 | 22 (branch model) |
| Lysin | 120 | 17 | 32 | 11 | 15 | 11 (branch model), 9 (branch-site model) |
| APOL1–4 | 312 | 25 | 3 | 12 | 12 | 5 (branch model) 2 (branch-site model) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steffen, R.; Ogoniak, L.; Grundmann, N.; Pawluchin, A.; Soehnlein, O.; Schmitz, J. paPAML: An Improved Computational Tool to Explore Selection Pressure on Protein-Coding Sequences. Genes 2022, 13, 1090. https://doi.org/10.3390/genes13061090
Steffen R, Ogoniak L, Grundmann N, Pawluchin A, Soehnlein O, Schmitz J. paPAML: An Improved Computational Tool to Explore Selection Pressure on Protein-Coding Sequences. Genes. 2022; 13(6):1090. https://doi.org/10.3390/genes13061090
Chicago/Turabian StyleSteffen, Raphael, Lynn Ogoniak, Norbert Grundmann, Anna Pawluchin, Oliver Soehnlein, and Jürgen Schmitz. 2022. "paPAML: An Improved Computational Tool to Explore Selection Pressure on Protein-Coding Sequences" Genes 13, no. 6: 1090. https://doi.org/10.3390/genes13061090