
Transcriptome and Differentially Expressed Gene Profiles in Mycelium, Primordium and Fruiting Body Development in Stropharia rugosoannulata

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of S. rugosoannulata Materials at Different Developmental Stages
2.2. Total RNA Isolation, cDNA Library Preparation and Illumina Sequencing
2.3. Read Quality Control and Mapping
2.4. Differential Expression Analysis and Functional Enrichment
2.5. Validation of Gene Expression by Quantitative Real-Time Polymerase Chain Reaction (qRT–PCR)
2.6. Statistical Analysis
3. Results
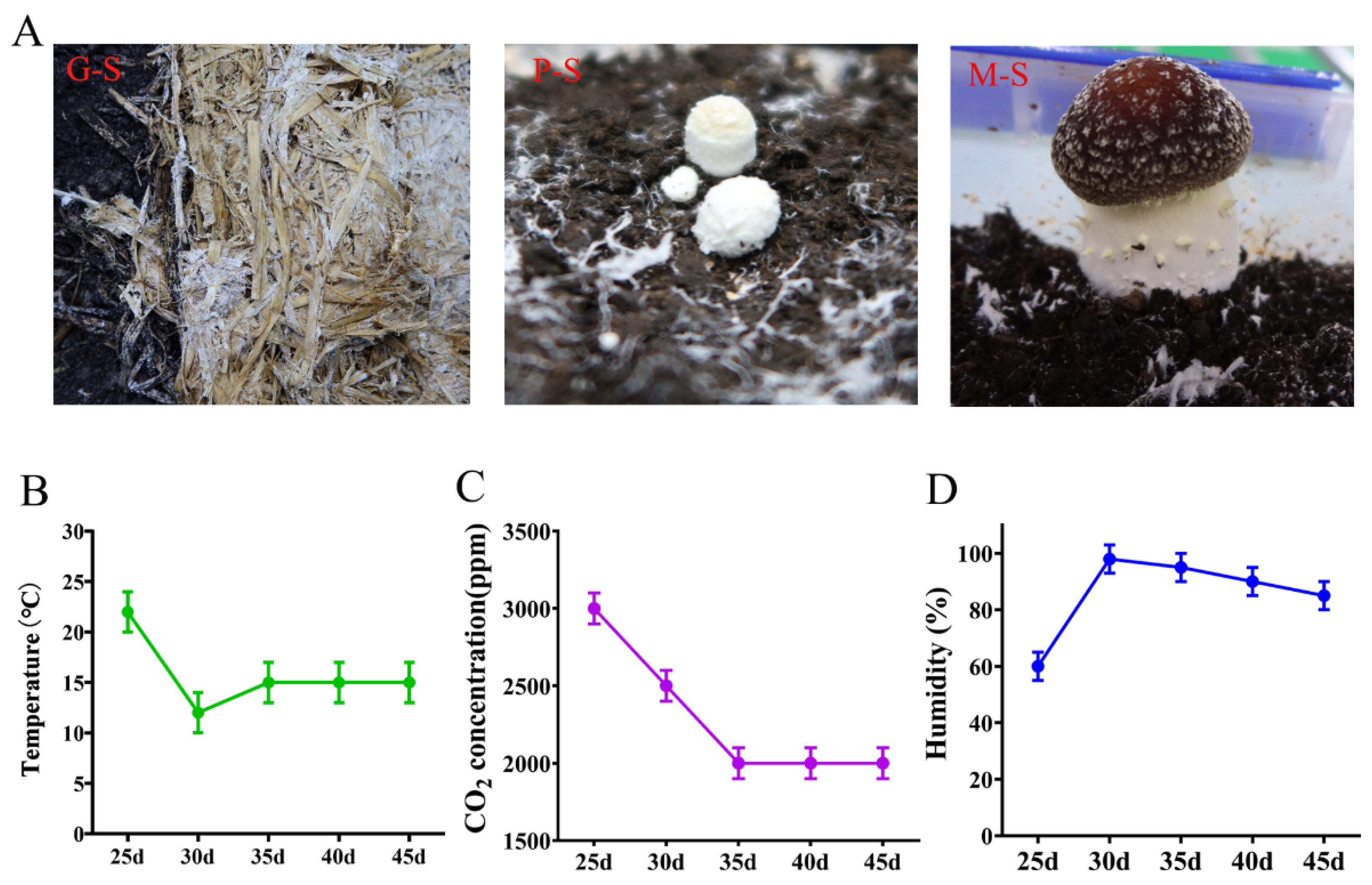
3.1. Analysis of the Morphological Features of S. rugosoannulata
3.2. Global Transcriptomic Analysis of S. rugosoannulata and Identification of DEGs
3.3. GO Enrichment and KEGG Pathway Analysis for DEGs
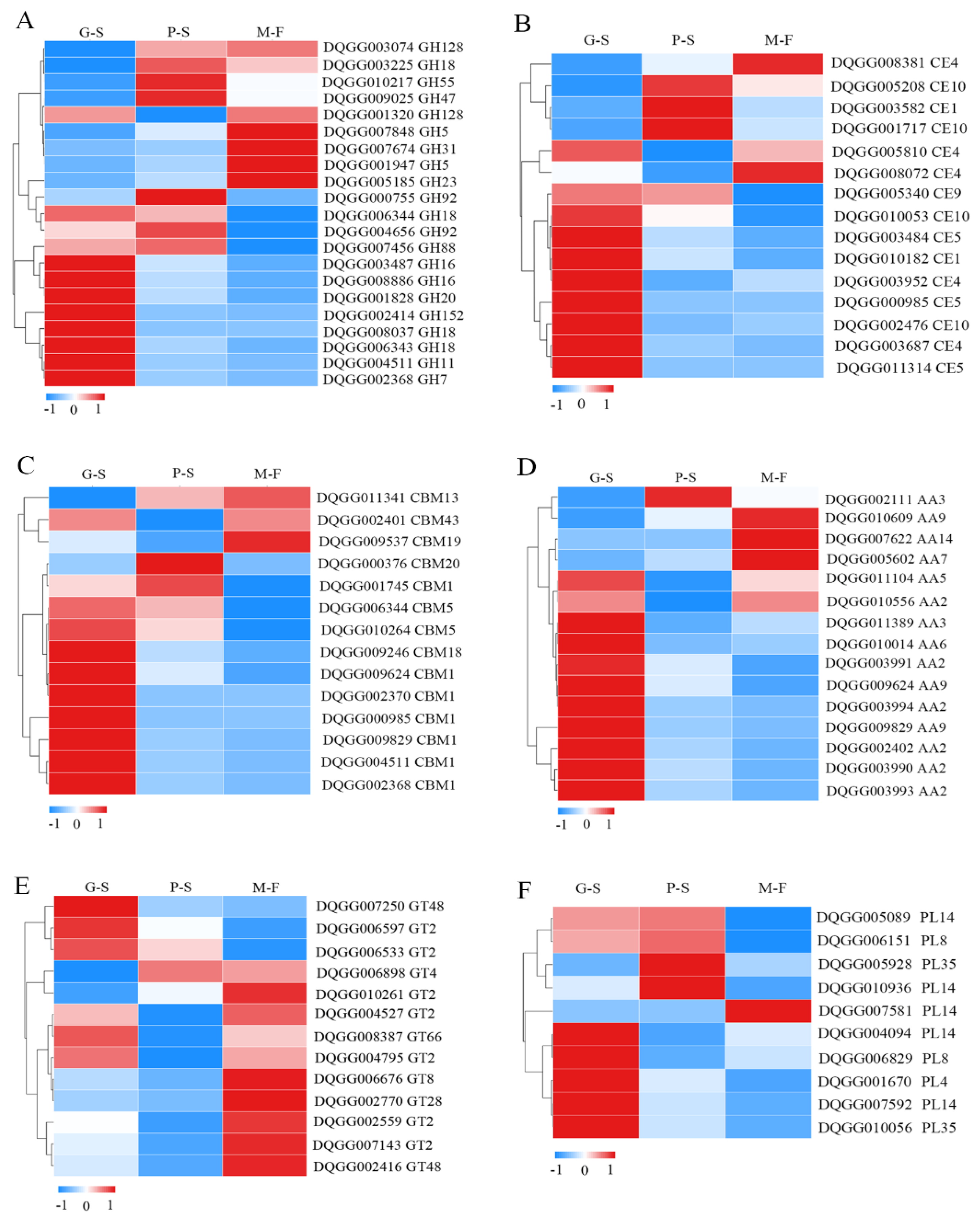
3.4. Differential Expression of Carbohydrate Enzyme Genes in Different Growth Developmental Stages
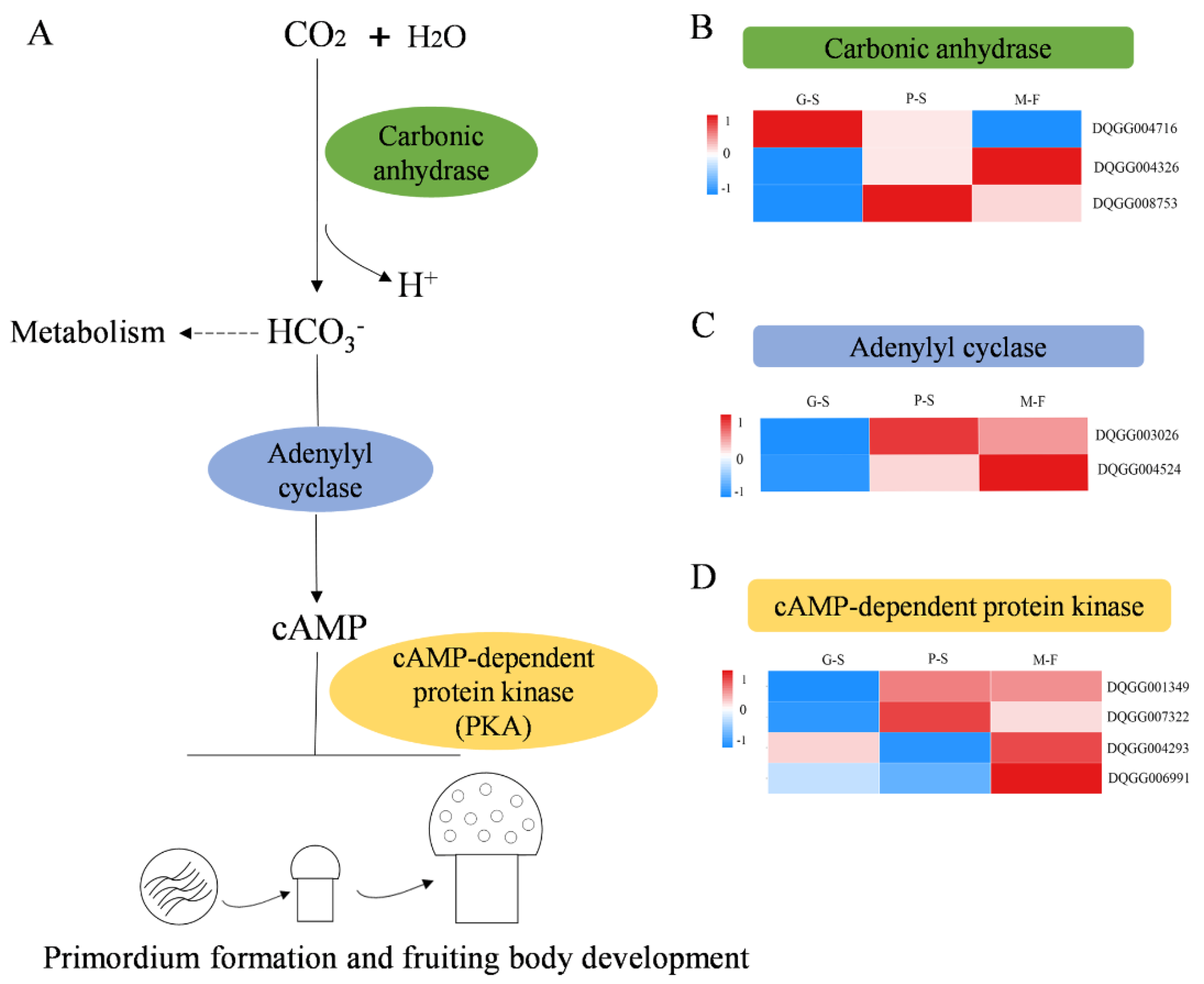
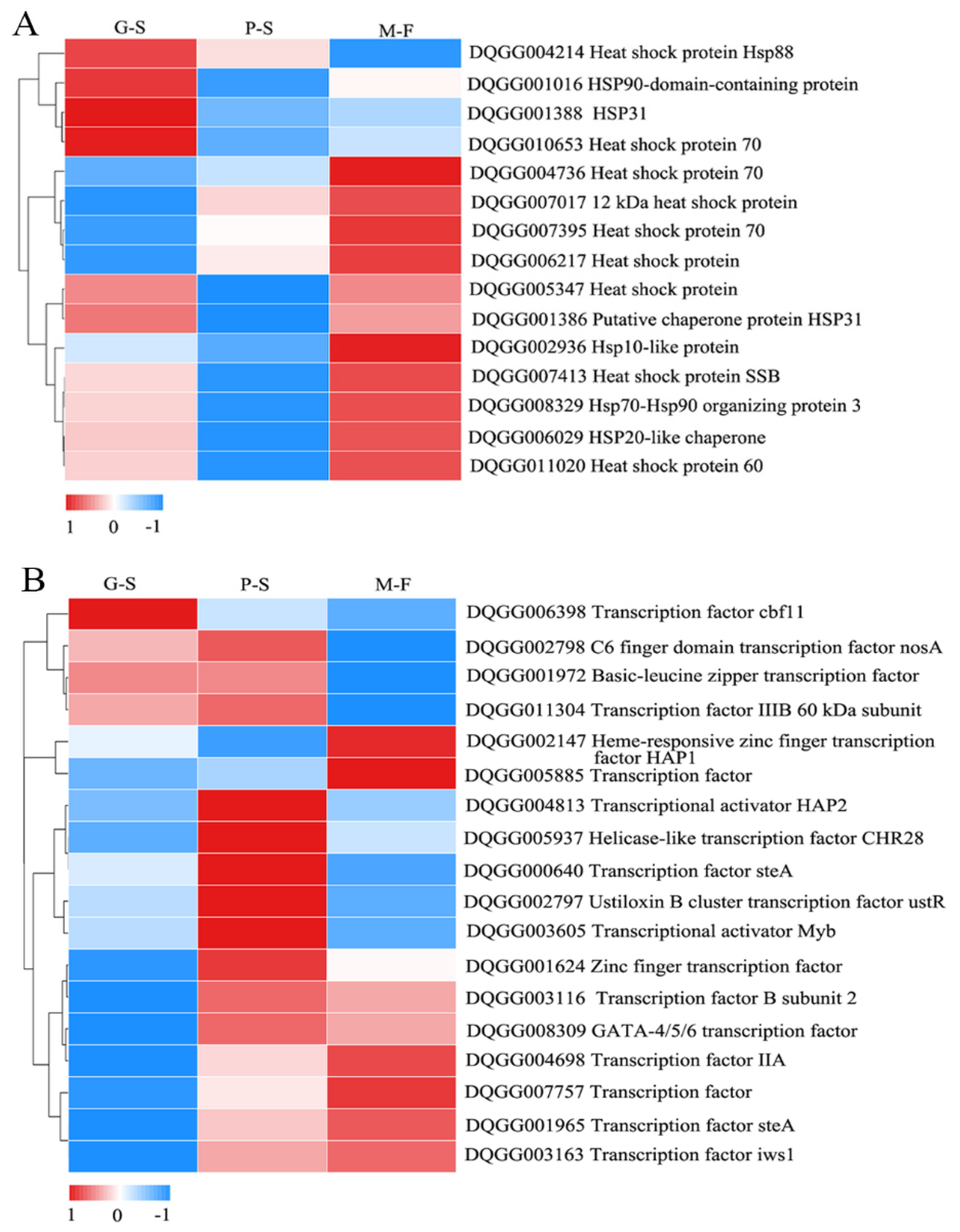
3.5. DEGs Related to Primordium Formation during Growth Developmental Stages
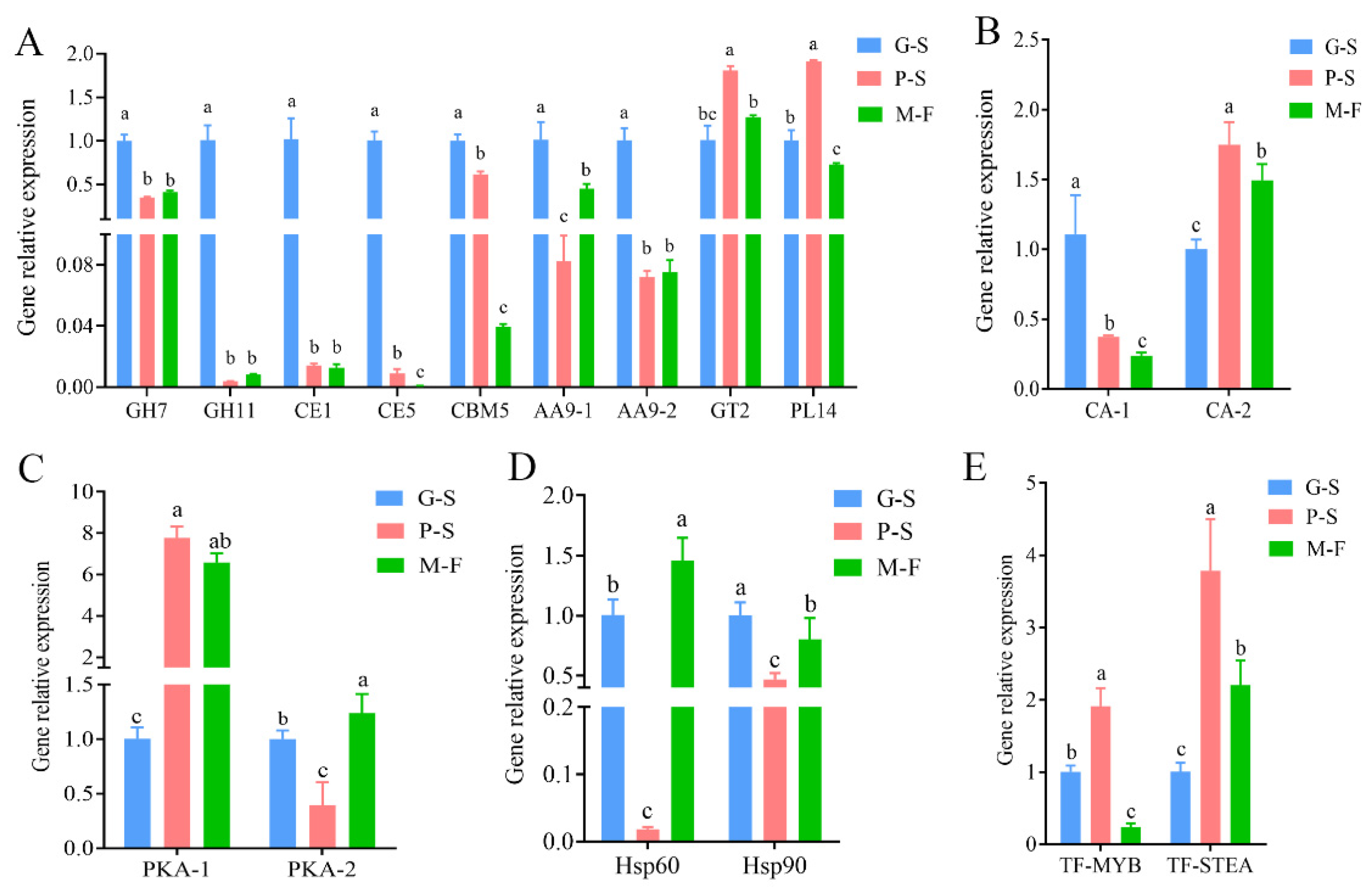
3.6. Validation of Transcriptomics Data by RT–qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Szudyga, K. Stropharia rugoso-annulata. In The Biology and Cultivation of Edible Mushrooms; Chang, S.T., Hayes, W.A., Eds.; Academic Press: Orlando, FL, USA, 1978; pp. 559–571. [Google Scholar]
- Yan, P.S.; Jiang, J.H. Preliminary research of the RAPD molecular marker-assisted breeding of the edible basidiomycete Stropharia rugoso-annulata. World J. Microb. Biot. 2005, 21, 559–563. [Google Scholar] [CrossRef]
- de Oliveira, J.B.H.; Pereira, P.R.C.; dos Santos, V.S.; Ferreira, J.M.; Dutra, J.C.V. Stropharia. In Benefcial Microbes in Agro-Ecology: Bacteria and Fungi; Amaresan, N., Kumar, S.M., Annapurna, K., Kumar, K., Sankranarayanan, A., Eds.; Academic Press: New York, NY, USA, 2020; p. 752. [Google Scholar]
- Zhang, Y.; Ni, J.; Yang, J.; Zhang, T.; Xie, D. Citrus stand ages regulate the fraction alteration of soil organic carbon under a citrus/stropharua rugodo-annulata intercropping system in the three gorges reservoir area, china. Environ. Sci. Pollut. Res. 2017, 24, 18363–18371. [Google Scholar] [CrossRef]
- Patyshakuliyeva, A.; Jurak, E.; Kohler, A.; Baker, A.; Battaglia, E.; de Bruijn, W.; Challen, M.P.; Coutinho, P.M.; Eastwood, D.C.; Gruben, B.S.; et al. Carbohydrate utilization and metabolism is highly differentiated in Agaricus bisporus. BMC Genom. 2013, 14, 663. [Google Scholar] [CrossRef] [Green Version]
- Rytioja, J.; Hildén, K.; Yuzon, J.; Hatakka, A.; de Vries, R.P.; Mäkelä, M.R. Plant-polysaccharide-degrading enzymes from basidiomycetes. Microbiol. Mol. Biol. Rev. 2014, 78, 614–649. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.F.; Zhu, J.X.; Wang, Q.J.; Gao, X.M.; Jiang, S.X. Breeding of Stropharia rugosoannulata “Shan nong qiu gai No. 3”. Mycosystema 2020, 39, 977–982. [Google Scholar]
- Sakamoto, Y. Influences of environmental factors on fruiting body induction, development and maturation in mushroom-forming fungi. Fungal Biol. Rev. 2018, 32, 236–248. [Google Scholar] [CrossRef]
- Kinugawa, K.; Suzuki, A.; Takamatsu, Y.; Kato, M.; Tanaka, K. Effects of concentrated carbon dioxide on the fruiting of several cultivated basidiomycetes (II). Mycoscience 1994, 35, 345–352. [Google Scholar] [CrossRef]
- Sietsma, J.H.; Rast, D.; Wessels, D.J. The effect of carbon dioxide on fruiting and on the degradation of a cell-wall glucan in Schizophyhm commune. J. Gen. Microbiol. 1977, 102, 385–389. [Google Scholar] [CrossRef] [Green Version]
- Raudaskoski, M.; Salonen, M. Interrelationships between vegetative development and basidiocarp initiation. In The Ecology and Physiology of the Fungal Mycelium; Jennings, D.H., Rayner, A.D.M., Eds.; Cambridge University Press: Cambridge, UK, 1984; pp. 291–322. [Google Scholar]
- Stamets, P. Growing Gourmet and Medicinal Mushrooms, 3rd ed.; Ten Speed Press: Berkeley, CA, USA, 2000; p. 2. [Google Scholar]
- Nakazawa, T.; Miyazaki, Y.; Kaneko, S.; Shishido, K. Stimulative effects of light and a temperature downshift on transcriptional expressions of developmentally regulated genes in the initial stages of fruiting-body formation of the basidiomycetous mushroom Lentinula edodes. FEMS Microbiol. Lett. 2008, 289, 67–71. [Google Scholar] [CrossRef] [Green Version]
- Morin, E.; Kohler, A.; Baker, A.R.; Foulongne-Oriol, M.; Lombard, V.; Nagy, L.G.; Ohm, R.A.; Patyshakuliyeva, A.; Brun, A.; Aerts, A.L.; et al. Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proc. Natl. Acad. Sci. USA 2012, 109, 17501–17506. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, Y.; Ando, A.; Tamai, Y.; Miura, K.; Yajima, T. Protein expressions during fruit body induction of Flammulina velutipes under reduced temperature. Mycol. Res. 2002, 106, 222–227. [Google Scholar] [CrossRef]
- Leatham, G.F.; Stahmann, M.A. Effect of light and aeration on fruiting of Lentinula edodes. Trans. Br. Mycol. Soc. 1987, 88, 9–20. [Google Scholar] [CrossRef]
- Kitamoto, Y.; Takahashi, M.; Kasai, Z. Light-induced formation of fruit-bodies in a basidiomycete, Favolus arcularius (FR.) AMES. Plant Cell Physiol. 1968, 9, 797–805. [Google Scholar]
- Tsusué, Y.M. Experimental control of fruit-body formation in Coprinus macrohizus. Dev. Growth Differ. 1969, 11, 164–178. [Google Scholar] [CrossRef]
- Hao, H.B.; Zhao, J.; Yang, H.; Zhang, J.X.; Wei, Y.H.; Kuai, B.K.; Zhang, J.J.; Chen, H. Comprehensive evaluation of main agronomic traits and nutritional components of different stropharia rugosoannulata strains. Acta Edulis Fungi 2022, 29, 41–49. [Google Scholar]
- Ohm, R.A.; de Jong, J.F.; Lugones, L.G.; Aerts, A.; Kothe, E.; Stajich, J.E.; De Vries, R.P.; Record, E.; Levasseur, A.; Baker, S.E.; et al. Genome sequence of the model mushroom Schizophyllum commune. Nat. Biotechnol. 2010, 28, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.J.; Wang, M.; Huang, J.; Yin, Y.L.; Chen, Y.J.; Jiang, S.; Jin, Y.X.; Lan, X.Q.; Wong, B.H.; Liang, Y.; et al. Deep insight into the Ganoderma lucidum by comprehensive analysis of its transcriptome. PLoS ONE 2012, 7, e44031. [Google Scholar] [CrossRef]
- Muraguchi, H.; Umezawa, K.; Niikura, M.; Yoshida, M.; Kozaki, T.; Ishii, K.; Sakai, K.; Shimizu, M.; Nakahori, K.; Sakamoto, Y.; et al. Strand-specific RNA-Seq analyses of fruiting body development in Coprinopsis cinerea. PLoS ONE 2015, 10, e0141586. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.J.; Baek, J.H.; Lee, S.; Kim, C.; Rhee, H.; Kim, H.; Seo, J.S.; Park, H.R.; Yoon, D.E.; Nam, J.Y.; et al. Whole genome and global gene expression analyses of the model mushroom Flammulina velutipes reveal a high capacity for lignocellulose degradation. PLoS ONE 2014, 9, e93560. [Google Scholar] [CrossRef]
- Tao, Y.; van Peer, A.F.; Chen, B.; Chen, Z.; Zhu, J.; Deng, Y.; Jiang, Y.J.; Li, S.J.; Wu, T.J.; Xie, B.G. Gene expression profiling reveals large regulatory switches between succeeding stipe stages in Volvariella volvacea. PLoS ONE 2014, 9, e97789. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Gong, Y.; Cai, Y.; Liu, W.; Zhou, Y.; Xiao, Y.; Xu, Z.Y.; Liu, Y.; Lei, X.Y.; Wang, G.Z.; et al. Genome sequence of the edible cultivated mushroom Lentinula edodes (Shiitake) reveals insights into lignocellulose degradation. PLoS ONE 2016, 11, e0160336. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Q.; Li, Q.P.; Qian, Z.M.; Zhang, X.L.; Li, K.; Li, W.J.; Dong, C.H. Developmental transcriptomics of Chinese cordyceps reveals gene regulatory network and expression profiles of sexual development-related genes. BMC Genom. 2019, 20, 337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hao, H.; Chen, M.; Wang, H.; Feng, Z.; Chen, H. Hydrogen-rich water alleviates the toxicities of different stresses to mycelial growth in Hypsizygus marmoreus. AMB Express 2017, 7, 107. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Brink, J.V.D.; Vries, R.P.D. Fungal enzyme sets for plant polysaccharide degradation. Appl. Microbiol. Biotechnol. 2011, 91, 1477–1492. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.Y.; Tong, Z.J.; Li, Y.N.; Yan, J.J.; Zhao, S.; Yao, S.; Yu, L.B.; Xie, B.G. CA gene superfamily and its expressions in response to CO2 in Flammulina filiformis. Mycosystema 2019, 38, 2249–2257. [Google Scholar]
- Martin, R.; Pohlers, S.; Mühlschlegel, F.A.; Kurzai, O. CO2 sensing in fungi: At the heart of metabolic signaling. Curr. Genet. 2017, 63, 965–972. [Google Scholar] [CrossRef]
- Gong, S.; Chen, C.; Zhu, J.X.; Qi, G.G.; Jinag, S.X. Effects of wine-cap Stropharia cultivation on soil nutrients and bacterial communities in forestlands of northern China. PeerJ 2018, 6, e5741. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Jia, L.; Xu, F.; Meng, F.; Deng, P.; Fan, K.; Liu, X.N. Characteristics of Se-enriched mycelia by Stropharia rugoso-annulata and its antioxidant activities in vivo. Biol. Trace Elem. Res. 2009, 131, 81–89. [Google Scholar] [CrossRef]
- Li, F.; Chen, L.D.; Ai, L.Y.; Liu, Y.C.; Yan, M.; Sun, S.J. Comparative transcriptomics analyses of Pleurotus eryngii at different developmental stages. Mycosystema 2018, 37, 1586–1597. [Google Scholar]
- Hao, H.B.; Zhang, J.J.; Wang, H.; Wang, Q.; Chen, M.J.; Juan, J.X.; Feng, Z.Y.; Chen, H. Comparative transcriptome analysis reveals potential fruiting body formation mechanisms in Morchella importuna. AMB Express 2019, 9, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabel, M.A.; Jurak, E.; Mäkelä, M.R.; de Vries, R.P. Occurrence and function of enzymes for lignocellulose degradation in commercial agaricus bisporus cultivation. Appl. Microbiol. Biotechnol. 2017, 101, 4363–4369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrik, A.; Jarmo, S.; Pedro, M.; Coutinho, M.S.; Kallas, A.; Djerbi, S.; Nilsson, P.; Denman, S.; Amini, B.; Sterky, F.; et al. Carbohydrate-active enzymes involved in the secondary cell wall biogenesis in hybrid aspen. Plant Physiol. 2005, 137, 983–997. [Google Scholar]
- Zhang, J.; Ren, A.; Chen, H.; Zhao, M.; Shi, L.; Chen, M.; Wang, H.; Feng, Z.Y. Transcriptome analysis and its application in identifying genes associated with fruiting body development in basidiomycete Hypsizygus marmoreus. PLoS ONE 2015, 10, e0123025. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.P.; Lian, L.D.; Guo, L.X.; Wang, W.; Chen, B.Z.; van Peer, A.F.; Li, S.J.; Wu, T.J.; Xie, B.G. The Accordant Trend of Both Parameters (rgs Expression and cAMP Content) Follows the Pattern of Development of Fruiting Body in Volvariella volvacea. Curr. Microbiol. 2015, 71, 579–584. [Google Scholar] [CrossRef]
- Fu, Y.P.; Liang, Y.; Dai, Y.T.; Yang, C.T.; Duan, M.Z.; Zhang, Z.; Hu, S.N.; Zhang, Z.W.; Li, Y. De Novo sequencing and transcriptome analysis of Pleurotus eryngii subsp. Tuoliensis (Bailinggu) mycelia in response to cold stimulation. Molecules 2016, 21, 560. [Google Scholar]
- Gulshan, K.; Moye-Rowley, W.S. Multidrug resistance in fungi. Eukaryot. Cell 2007, 6, 1933–1942. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.W.; Butchko, R.A.E.; Busman, M.; Proctor, R.H. The fusarium verticillioides FUM gene cluster encodes a Zn (II)2Cys6 protein that affects FUM gene expression and fumonisin production. Eukaryot. Cell 2007, 6, 1210–1218. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.P.; Cao, H.J.; Zhang, L.L.; Huang, P.Y.; Lin, F.C. Systematic analysis of Zn2Cys6 transcription factors required for development and pathogenicity by high-throughput gene knockout in the rice blast fungus. PLoS Pathog. 2014, 10, e1004432. [Google Scholar] [CrossRef]
- Yu, Y.T.; Wu, Z.; Lu, K.; Bi, C.; Liang, S.; Wang, X.F.; Zhang, D.P. Overexpression of the MYB37 transcription factor enhances abscisic acid sensitivity, and improves both drought tolerance and seed productivity in arabidopsis thaliana. Plant Mol. Biol. 2016, 90, 267–279. [Google Scholar] [CrossRef] [Green Version]
- Vallim, M.A.; Miller, K.Y.; Miller, B.L. Aspergillus SteA (sterile12-like) is a homeodomain-C2/H2−Zn+2 finger transcription factor required for sexual reproduction. Mol. Microbiol. 2000, 36, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Soukup, A.A.; Farnoodian, M.; Berthier, E.; Keller, N.P. NosA, a transcription factor important in Aspergillus fumigatus stress and developmental response, rescues the germination defect of a laeA deletion. Fungal Genet. Biol. 2012, 49, 857–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaisne, M.; Bécam, A.M.; Verdière, J.; Herbert, C.J. A’natural’mutation in Saccharomyces cerevisiae strains derived from S288c affects the complex regulatory gene HAP1 (CYP1). Curr. Genet. 1999, 36, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Laverriere, A.C.; MacNeill, C.; Mueller, C.; Poelmann, R.E.; Burch, J.B.; Evans, T. GATA-4/5/6, a subfamily of three transcription factors transcribed in developing heart and gut. J. Biol. Chem. 1994, 269, 23177–23184. [Google Scholar] [CrossRef]
- Colot, H.; Park, G.; Turner, G.; Ringelberg, C.; Crew, C.; Litvinkova, L.; Weiss, R.L.; Borkovich, K.A.; Dunlap, J.C. A high-throughput gene knockout procedure for neurospora reveals functions for multiple transcription factors. Proc. Natl. Acad. Sci. USA 2006, 103, 10352–10357. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, H.; Zhang, J.; Wang, Q.; Huang, J.; Juan, J.; Kuai, B.; Feng, Z.; Chen, H. Transcriptome and Differentially Expressed Gene Profiles in Mycelium, Primordium and Fruiting Body Development in Stropharia rugosoannulata. Genes 2022, 13, 1080. https://doi.org/10.3390/genes13061080
Hao H, Zhang J, Wang Q, Huang J, Juan J, Kuai B, Feng Z, Chen H. Transcriptome and Differentially Expressed Gene Profiles in Mycelium, Primordium and Fruiting Body Development in Stropharia rugosoannulata. Genes. 2022; 13(6):1080. https://doi.org/10.3390/genes13061080
Chicago/Turabian StyleHao, Haibo, Jinjing Zhang, Qian Wang, Jianchun Huang, Jiaxiang Juan, Benke Kuai, Zhiyong Feng, and Hui Chen. 2022. "Transcriptome and Differentially Expressed Gene Profiles in Mycelium, Primordium and Fruiting Body Development in Stropharia rugosoannulata" Genes 13, no. 6: 1080. https://doi.org/10.3390/genes13061080