
Central Core Disease: Facial Weakness Differentiating Biallelic from Monoallelic Forms

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients Clinical Evaluation
2.2. Molecular Analysis
2.3. Data Analysis
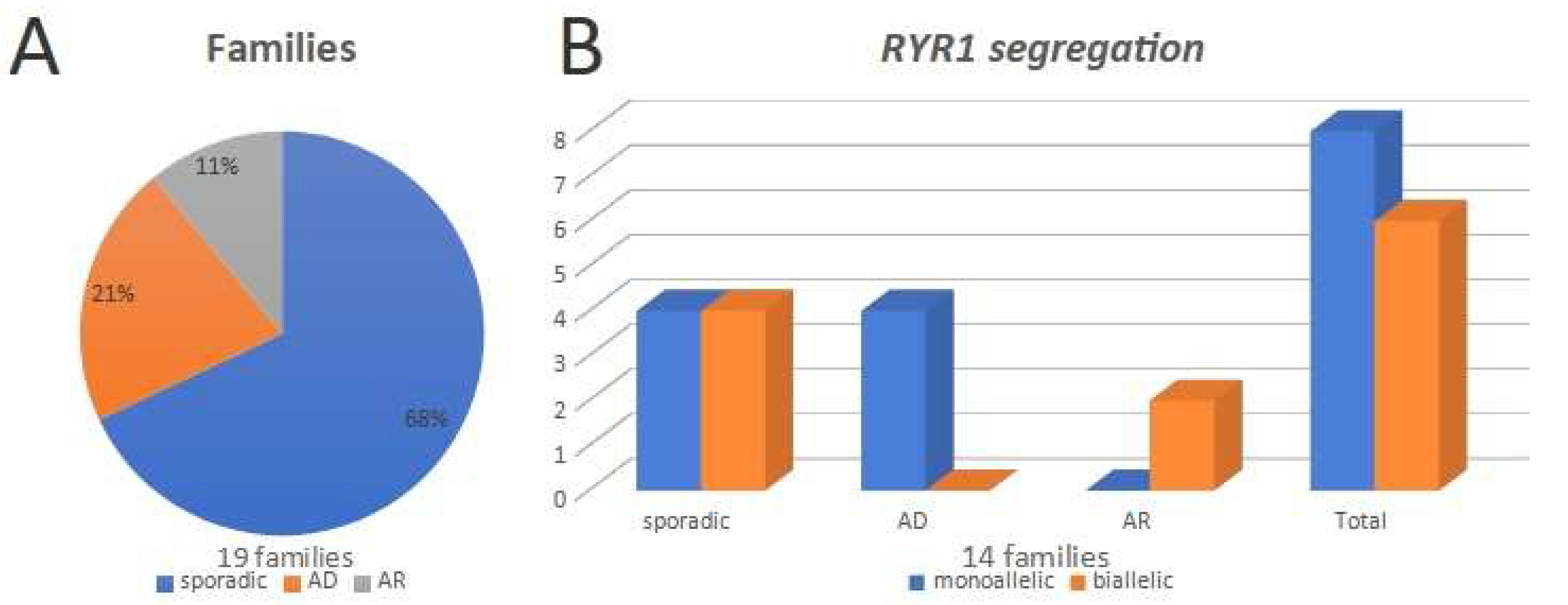
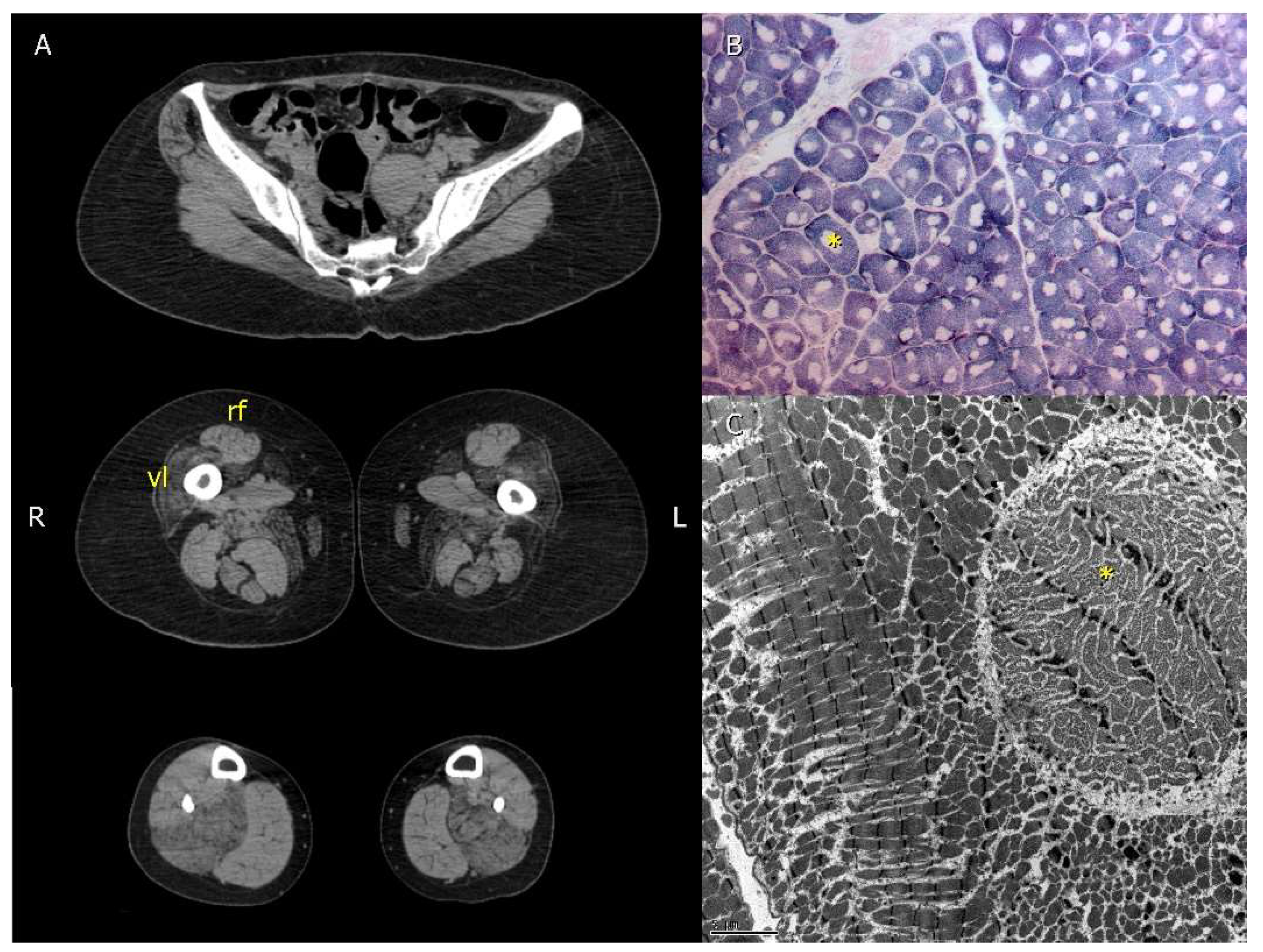
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dubowitz, V.; Brooke, M.H. Muscle Biopsy: A Modern Approach; W. B. Saunders Company Ltd.: Philadelphia, PA, USA, 1973. [Google Scholar]
- Dubowitz, V.; Sewry, C.A.; Oldfors, A. Muscle Biopsy: A Practical Approach, 5th ed.; Elsevier Ltd.: Beijing, China, 2021. [Google Scholar]
- Karpati, G.; Hilton-Jones, D.; Bushby, K.; Griggs, R.C. Disorders of Voluntary Muscle, 10th ed.; Cambridge University Press: Cambridge, UK, 2010. [Google Scholar]
- Jungbluth, H.; Sewry, C.A.; Muntoni, F. Core Myopathies. Semin. Pediatr. Neurol. 2011, 18, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Amburgey, K.; Bailey, A.; Hwang, J.H.; Tarnopolsky, M.A.; Bonnemann, C.G.; Medne, L.; Mathews, K.D.; Collins, J.; Daube, J.R.; Wellman, G.P.; et al. Genotype-Phenotype Correlations in Recessive RYR1-Related Myopathies. Orphanet J. Rare Dis. 2013, 8, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotta, A.; Carvalho, E.; Da-Cunha-Júnior, A.L.; Valicek, J.; Navarro, M.M.; Junior, S.B.; da Silveira, E.B.; Lima, M.I.; Cordeiro, B.A.; Cauhi, A.F.; et al. Muscle Biopsy Essential Diagnostic Advice for Pathologists. Surg. Exp. Pathol. 2021, 4, 3. [Google Scholar] [CrossRef]
- Kossugue, P.M.; Paim, J.F.; Navarro, M.M.; Silva, H.C.; Pavanello, R.C.M.; Gurgel-Giannetti, J.; Zatz, M.; Vainzof, M. Central Core Disease Due to Recessive Mutations in RYR1 Gene: Is It More Common than Described? Muscle Nerve 2007, 35, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Leão, L.G.; Souza, L.S.; Nogueira, L.; Pavanello, R.D.C.M.; Gurgel-Giannetti, J.; Reed, U.C.; Oliveira, A.S.B.; Cuperman, T.; Cotta, A.; Paim, J.F.; et al. Dominant or Recessive Mutations in the RYR1 Gene Causing Central Core Myopathy in Brazilian Patients. Acta Myol. 2020, 39, 274–282. [Google Scholar] [CrossRef]
- Gambelli, S.; Malandrini, A.; Berti, G.; Gaudiano, C.; Zicari, E.; Brunori, P.; Perticoni, G.; Orrico, A.; Galli, L.; Sorrentino, V.; et al. Inheritance of a Novel RYR1 Mutation in a Family with Myotonic Dystrophy Type 1. Clin. Genet. 2007, 71, 93–94. [Google Scholar] [CrossRef] [PubMed]
- Samões, R.; Oliveira, J.; Taipa, R.; Coelho, T.; Cardoso, M.; Gonçalves, A.; Santos, R.; Melo Pires, M.; Santos, M. RYR1-Related Myopathies: Clinical, Histopathologic and Genetic Heterogeneity Among 17 Patients from a Portuguese Tertiary Centre. J. Neuromuscul. Dis. 2017, 4, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Abath Neto, O.; Moreno, C.d.A.M.; Malfatti, E.; Donkervoort, S.; Böhm, J.; Guimarães, J.B.; Foley, A.R.; Mohassel, P.; Dastgir, J.; Bharucha-Goebel, D.X.; et al. Common and Variable Clinical, Histological, and Imaging Findings of Recessive RYR1 -Related Centronuclear Myopathy Patients. Neuromuscul. Disord. 2017, 27, 975–985. [Google Scholar] [CrossRef]
- Cotta, A.; Paim, J.F.; Pavanello, R.D.C.M.; Nogueira, L.; Leão, L.G.; Xavier-Neto, R.; Navarro, M.M.; Carvalho, E.; Valicek, J.; Silveira, E.B.; et al. Central Core Myopathy with Autophagy. Muscle Nerve 2017, 56, E8–E9. [Google Scholar] [CrossRef]
- Monnier, N.; Romero, N.B.; Lerale, J.; Landrieu, P.; Nivoche, Y.; Fardeau, M.; Lunardi, J. Familial and Sporadic Forms of Central Core Disease Are Associated with Mutations in the C-terminal Domain of the Skeletal Muscle Ryanodine Receptor. Hum. Mol. Genet. 2001, 10, 2581–2592. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.; Haan, E.; Jungbluth, H.; Sewry, C.; North, K.; Muntoni, F.; Kuntzer, T.; Lamont, P.; Bankier, A.; Tomlinson, P.; et al. Principal Mutation Hotspot for Central Core Disease and Related Myopathies in the C-terminal Transmembrane Region of the RYR1 Gene. Neuromuscul. Disord. 2003, 13, 151–157. [Google Scholar] [CrossRef]
- Chae, J.H.; Vasta, V.; Cho, A.; Lim, B.C.; Zhang, Q.; Eun, S.H.; Hahn, S.H. Utility of next Generation Sequencing in Genetic Diagnosis of Early Onset Neuromuscular Disorders. J. Med. Genet. 2015, 52, 208–216. [Google Scholar] [CrossRef]
- Rueffert, H.; Olthoff, D.; Deutrich, C.; Meinecke, C.D.; Froster, U.G. Mutation Screening in the Ryanodine Receptor 1 Gene (RYR1) in Patients Susceptible to Malignant Hyperthermia Who Show Definite IVCT Results: Identification of Three Novel Mutations. Acta Anaesthesiol. Scand. 2002, 46, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Gillard, E.F.; Otsu, K.; Fujii, J.; Khanna, V.K.; de Leon, S.; Derdemezi, J.; Britt, B.A.; Duff, C.L.; Worton, R.G.; MacLennan, D.H. A Substitution of Cysteine for Arginine 614 in the Ryanodine Receptor Is Potentially Causative of Human Malignant Hyperthermia. Genomics 1991, 11, 751–755. [Google Scholar] [CrossRef]
- Monnier, N.; Marty, I.; Faure, J.; Castiglioni, C.; Desnuelle, C.; Sacconi, S.; Estournet, B.; Ferreiro, A.; Romero, N.; Laquerriere, A.; et al. Null Mutations Causing Depletion of the Type 1 Ryanodine Receptor (RYR1) Are Commonly Associated with Recessive Structural Congenital Myopathies with Cores. Hum. Mutat. 2008, 29, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Xie, Y.; Ma, J.; Luo, X.; Nie, P.; Zuo, Z.; Lahrmann, U.; Zhao, Q.; Zheng, Y.; Zhao, Y.; et al. IBS: An Illustrator for the Presentation and Visualization of Biological Sequences: Figure 1. Bioinformatics 2015, 31, 3359–3361. [Google Scholar] [CrossRef] [Green Version]
- North, K.N.; Wang, C.H.; Clarke, N.; Jungbluth, H.; Vainzof, M.; Dowling, J.J.; Amburgey, K.; Quijano-Roy, S.; Beggs, A.H.; Sewry, C.; et al. International Standard of Care Committee for Congenital Myopathies. Approach to the Diagnosis of Congenital Myopathies. Neuromuscul. Disord. 2014, 24, 97–116. [Google Scholar] [CrossRef] [Green Version]
- Gonorazky, H.D.; Bönnemann, C.G.; Dowling, J.J. The genetics of congenital myopathies. Handb. Clin. Neurol. 2018, 148, 549–564. [Google Scholar] [CrossRef]
- Norwood, F.L.M.; Harling, C.; Chinnery, P.F.; Eagle, M.; Bushby, K.; Straub, V. Prevalence of Genetic Muscle Disease in Northern England: In-Depth Analysis of a Muscle Clinic Population. Brain 2009, 132, 3175–3186. [Google Scholar] [CrossRef] [Green Version]
- Cuisset, J.-M.; Maurage, C.-A.; Carpentier, A.; Briand, G.; Thévenon, A.; Rouaix, N.; Vallée, L. [Muscle Biopsy in Children: Usefulness in 2012]. Intérêt de La Biopsie Musculaire Chez l’enfant En 2012. Rev. Neurol. (Paris) 2013, 169, 632–639. [Google Scholar] [CrossRef]
- Cotta, A.; Paim, J.F.; Carvalho, E.; Da-Cunha-Júnior, A.L.; Navarro, M.M.; Valicek, J.; Menezes, M.M.; Nunes, S.V.; Xavier-Neto, R.; Baptista Junior, S.; et al. The Relative Frequency of Common Neuromuscular Diagnoses in a Reference Center. Arq. Neuropsiquiatr. 2017, 75, 789–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafner, P.; Phadke, R.; Manzur, A.; Smith, R.; Jaiser, S.; Schutz, P.; Sewry, C.; Muntoni, F.; Pitt, M. Electromyography and Muscle Biopsy in Paediatric Neuromuscular Disorders—Evaluation of Current Practice and Literature Review. Neuromuscul. Disord. 2019, 29, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.J.; Razaqyar, M.S.; Witherspoon, J.W.; Lawal, T.A.; Mankodi, A.; Chrismer, I.C.; Allen, C.; Meyer, M.D.; Kuo, A.; Shelton, M.S.; et al. Novel Variants in Individuals with RYR1-Related Congenital Myopathies: Genetic, Laboratory, and Clinical Findings. Front. Neurol. 2018, 9, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, E.; Bonne, G.; Rivier, F.; Hamroun, D. The 2022 Version of the Gene Table of Neuromuscular Disorders (Nuclear Genome). Neuromuscul. Disord. 2021, 31, 1313–1357. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Zhang, S.; Hu, J.; Yuan, Y.; Wang, Z.; Da, Y.; Wu, S. Novel RYR1 Missense Mutations in Six Chinese Patients with Central Core Disease. Neurosci. Lett. 2014, 566, 32–35. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fam | Patient | Inherit. | Start | Age | Gender | Oligo. | Akinesia | Delay | Hypotonia | Bulbar | Resp. | Hip Disl. | Club Feet |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1.1 | D | 0 | 37 | M | NR | NR | Y | Y | N | N | N | N |
| 1 | 1.2 | D | 28 | 28 | F | N | N | Y | Y | N | N | N | N |
| 1 | 1.3 | D | 0 | 8 | M | N | N | Y | Y | N | N | N | N |
| 1 | 1.4 | D | 0 | 1 | M | N | N | Y | Y | N | N | N | N |
| 2 | 2.1 | D | 0 | 24 | F | NA | NA | Y | NA | NA | NA | NA | NA |
| 2 | 2.2 | D | 0 | 5 | M | N | N | Y | Y | Y | Y | N | N |
| 3 | 3.1 | D | 1 | 25 | F | NA | NA | N | N | NA | NA | N | N |
| 3 | 3.2 | D | NR | 47 | F | NA | NA | NA | NA | NA | NA | NA | NA |
| 4 | 4.1 | D | 2 | 18 | F | NR | NR | N | NR | NR | NR | N | N |
| 4 | 4.2 | D | 2 | 26 | M | NR | NR | NR | NR | NR | NR | N | N |
| 4 | 4.3 | D | 2 | 52 | F | NR | NR | NR | NR | NR | NR | N | N |
| 5 | 5.1 | R | 8 | 38 | M | N | N | N | N | N | N | N | N |
| 5 | 5.2 | R | 7 | 42 | F | N | N | N | N | N | N | N | Y |
| 6 | 6.1 | R | 0 | 1 | F | N | N | Y | Y | N | N | Y | N |
| 7 | 7.1 | S | 2 | 27 | M | NR | NR | N | N | N | N | N | N |
| 8 | 8.1 | S | 0 | 7 | F | Y | N | Y | Y | N | N | Y | Y |
| 9 | 9.1 | S | 0 | 17 | M | N | N | N | Y | N | N | N | N |
| 10 | 10.1 | S | 2 | 14 | M | NR | NR | NR | NR | NR | NR | NR | Y |
| 11 | 11.1 | S | 0 | 7 | M | NR | NR | N | N | N | N | N | N |
| 12 | 12.1 | S | 0 | 2 | F | Y | N | Y | Y | N | N | Y | N |
| 13 | 13.1 | S | 0 | 4 | M | N | N | Y | Y | N | N | Y | N |
| 14 | 14.1 | S | 0 | 9 | F | N | N | Y | Y | N | N | N | N |
| 15 | 15.1 | S | 0 | 4 | F | N | Y | Y | Y | N | N | Y | N |
| 16 | 16.1 | S | 0 | 5 | F | NR | NR | Y | Y | Y | NR | N | N |
| 17 | 17.1 | S | 0 | 41 | F | NR | NR | Y | NR | NR | NR | N | N |
| 18 | 18.1 | S | 0 | 9 | F | N | N | N | N | N | N | N | Y |
| 19 | 19.1 | S | 0 | 2 | F | N | N | Y | Y | Y | Y | N | N |
| F | P | I | Face | Oph | Pt | Dys | Hypo | Ax | Prox | Dist | Reflex | ENMG | CK (×) | Other Findings |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1.1 | D | N | N | N | N | Y | N | Y | N | A | M | 63 (<1×) | |
| 1 | 1.2 | D | N | N | N | N | N | N | Y | N | NR | M | 75 (<1×) | |
| 1 | 1.3 | D | N | N | N | N | Y | N | Y | N | A | M | 75 (<1×) | |
| 1 | 1.4 | D | N | N | N | N | N | Y | Y | N | A | NA | 102 (<1×) | |
| 2 | 2.1 | D | N | N | N | N | Y | N | Y | N | A | M | 267 (1.6×) | |
| 2 | 2.2 | D | N | N | N | N | Y | Y | Y | Y | A | M | 131 (<1×) | Knee contractures |
| 3 | 3.1 | D | N | N | N | N | Y | N | Y | N | NL | M | 74 (<1×) | Ankle contracture |
| 3 | 3.2 | D | N | N | N | N | N | NA | NA | NA | NA | NA | NA | |
| 4 | 4.1 | D | N | N | N | N | N | N | Y | N | NL | M | 156 (<1×) | Right calf atrophy, winged scapula |
| 4 | 4.2 | D | N | N | N | N | N | N | Y | N | NA | M | 62 (<1×) | |
| 4 | 4.3 | D | N | N | N | N | N | N | Y | N | NL | NA | 78 (<1×) | |
| 5 | 5.1 | R | Y | N | N | N | N | N | Y | N | Hypo | M | 189 (1×) | |
| 5 | 5.2 | R | N | N | N | N | N | N | Y | N | Hypo | M | 389 (2.8×) | Toe walking |
| 6 | 6.1 | R | Y | N | N | Y | N | Y | Y | N | Hypo | M | 52 (<1×) | |
| 7 | 7.1 | S | N | N | N | N | Y | N | Y | N | Hypo | M | 51 (<1×) | |
| 8 | 8.1 | S | Y | N | N | N | Y | N | Y | Y | A | M | 93 (<1×) | Gait with orthesis at age 5 |
| 9 | 9.1 | S | N | N | N | N | Y | N | Y | N | A | M | 50 (<1×) | No gait acquisition |
| 10 | 10.1 | S | N | N | N | N | N | Y | Y | N | Hypo | M | 39 (<1×) | Pes cavus |
| 11 | 11.1 | S | Y | N | N | N | N | Y | Y | Y | A | Mix | 79 (<1×) | |
| 12 | 12.1 | S | N | N | N | N | N | Y | Y | Y | A | M | 65 (<1×) | Pes cavus, knee contractures |
| 13 | 13.1 | S | Y | N | N | Y | Y | N | Y | N | Hypo | M | 36 (<1×) | Joint laxity |
| 14 | 14.1 | S | Y | N | Y | Y | Y | NA | Y | N | A | M | 33 (<1×) | Hydrocephalia, joint laxity |
| 15 | 15.1 | S | N | N | N | N | Y | N | Y | N | Hypo | NL | 49 (<1×) | Scoliosis, Left knee contracture |
| 16 | 16.1 | S | Y | N | Y | Y | Y | Y | Y | Y | A | M | 80 (<1×) | |
| 17 | 17.1 | S | N | N | N | N | N | N | Y | Y | NL | M | 293 (1.7×) | Pes cavus. Knee dislocation |
| 18 | 18.1 | S | N | N | N | Y | N | N | Y | Y | NL | M | 966 (4.3×) | Malignant hyperthermia episode. Small mouth |
| 19 | 19.1 | S | Y | N | N | Y | N | Y | Y | N | Hyper | NL | 67 (<1×) | Café-au-lait spots |
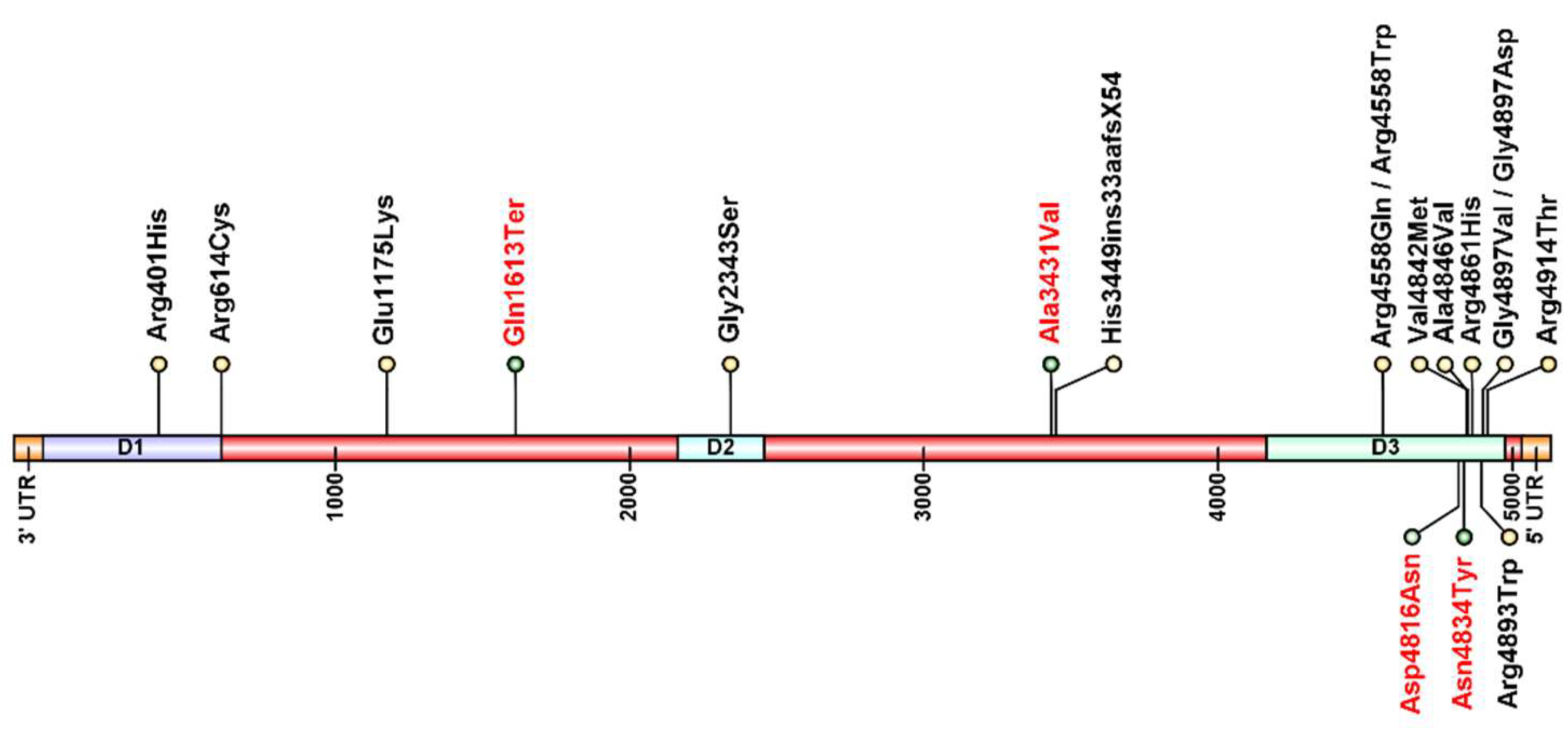
| Family | Patient | Inheritance | Variant | Protein Change | Exon | Reference |
|---|---|---|---|---|---|---|
| 1 | 1.1 | Dominant | c.14690G>T | p.Gly4897Val | 102 | Kossugue et al., 2007 [7]. Galleni-Leao et al., 2020 [8] |
| 2 | 2.1 | Dominant | c.14690G>A | p.Gly4897Asp | 102 | Amburguey et al., 2013 [5] |
| 3 | 3.1 | Dominant | c.14500A>T | p.Asn4834Tyr | 100 | Novel |
| 4 | 4.3 | Dominant | c.14446G>A | p.Asp4816Asn | 100 | Novel |
| 5 | 5.2 | Recessive | c.13673G>A | p.Arg4558Gln | 94 | Kossugue et al., 2007 [7] |
| c.14537C>T | p.Ala4846Val | 101 | Galleni-Leao et al., 2020 [8]. Gambelli et al., 2007 [9] | |||
| 6 | 6.1 | Recessive | c.7027G>A | p.Gly2343Ser | 43 | Samões et al., 2017 [10]. Abath-Neto et al., 2017 [11] |
| c.13672C>T | p.Arg4558Trp | 94 | Samões et al., 2017 [10]. Abath-Neto et al., 2017 [11] | |||
| c.14537C>T | p.Ala4846Val | 101 | Galleni-Leao et al., 2020 [8]. Gambelli et al., 2007 [9] | |||
| 7 | 7.1 | Sporadic | c.14677C>T | p.Arg4893Trp | 101 | Cotta et al., 2017 [12]. Galleni-Leão et al., 2020 [8] |
| 8 | 8.1 | Sporadic | c.14582G>A | p.Arg4861His | 101 | Monnier et al., 2001 [13] |
| 9 | 9.1 | Sporadic | c.14741G>C | p.Arg4914Thr | 102 | Galleni-Leão et al., 2020 [8]. Davis et al., 2003 [14] |
| 10 | 10.1 | Sporadic | c.14582G>A | p.Arg4861His | 101 | Galleni-Leão et al., 2020 [8]. Monnier et al., 2001 [13] |
| 11 | 11.1 | Sporadic | c.3523G>A | p.Glu1175Lys | 26 | Chae et al., 2015 [15] |
| c.4837C>T | p.Gln1613Ter | 33 | Novel | |||
| 12 | 12.1 | Sporadic | c.1202G>A | p.Arg401His | 12 | Rueffert et al., 2002 [16] |
| c.1840 C>T | p.Arg614Cys | 17 | Gillard et al., 1991 [17] | |||
| 13 | 13.1 | Sporadic | c.14292C>T | p.Ala3431Val | 68 | Novel |
| c.14582G>A | p.Arg4861His | 101 | Monnier et al., 2001 [13] | |||
| 14 | 14.1 | Sporadic | c.10348-6C>G | p.His3449ins33aafsX54 | - | Monnier et al., 2008 [18] |
| c.14524G>A | p.Val4842Met | 101 | Monnier et al., 2008 [18] | |||
| 15 | 15.1 | Sporadic | - | - | - | - |
| 16 | 16.1 | Sporadic | - | - | - | - |
| 17 | 17.1 | Sporadic | - | - | - | - |
| 18 | 18.1 | Sporadic | - | - | - | - |
| 19 | 19.1 | Sporadic | - | - | - | - |
| Monoallelic | Biallelic | ||||||
|---|---|---|---|---|---|---|---|
| Clinical Characteristic | Percentage | Yes | n | Percentage | Yes | n | p Value |
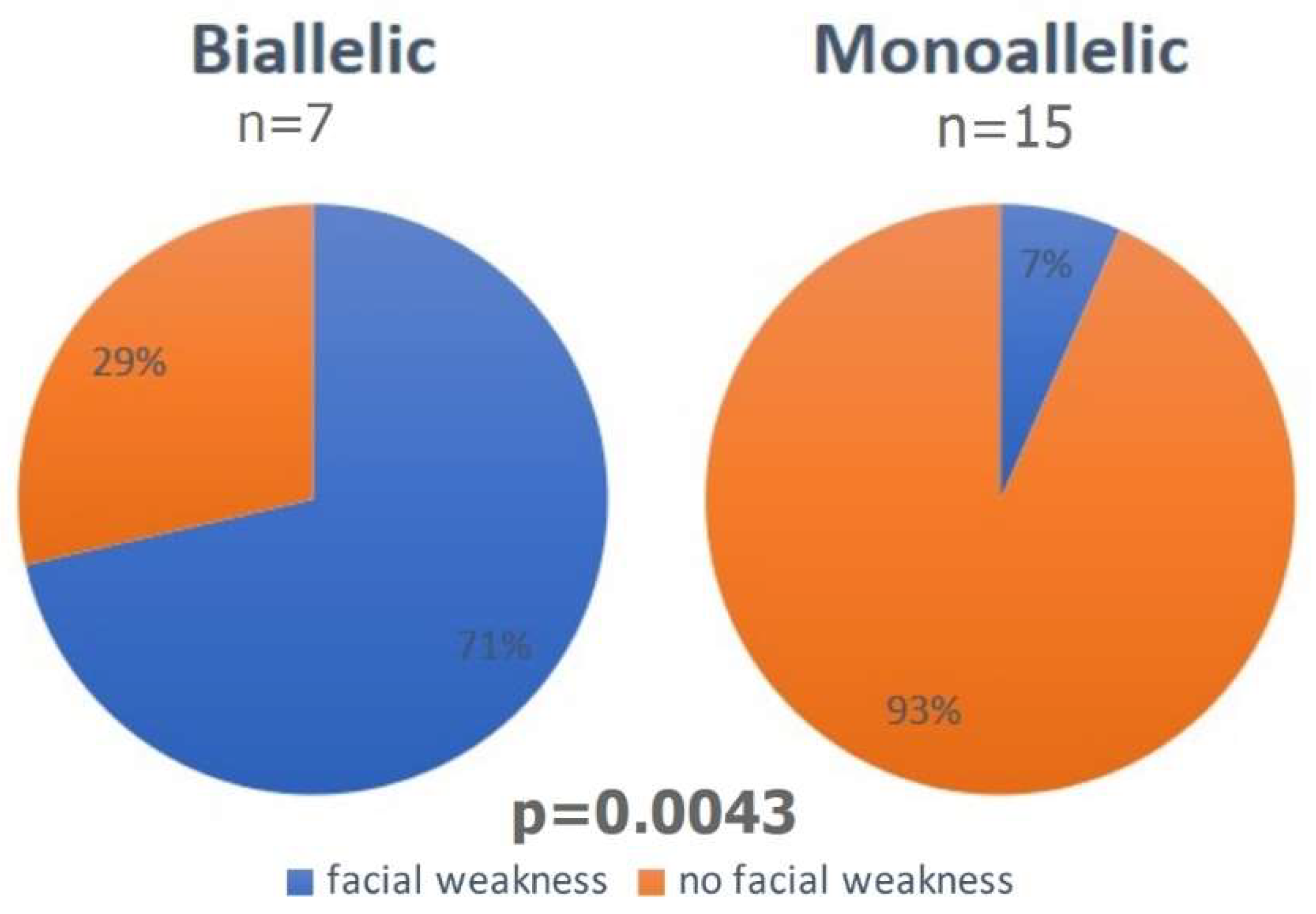
| Facial weakness | 6.7% | 1 | 15 | 71.0% | 5 | 7 | p = 0.0043 |
| Facial dysmorphism | 0% | 0 | 15 | 42.9% | 3 | 7 | p = 0.0227 |
| Axial weakness | 21.4% | 3 | 14 | 50.0% | 3 | 6 | p = 0.3027 |
| Distal weakness | 14.3% | 2 | 14 | 28.6% | 2 | 7 | p = 0.5743 |
| Hypotrophy | 53.3% | 8 | 15 | 28.6% | 2 | 7 | p = 0.3808 |
| Bulbar symptoms | 12.5% | 1 | 8 | 0% | 0 | 7 | p = 1.0000 |
| Congenital club feet | 15.4% | 2 | 13 | 14.3% | 1 | 7 | p = 1.0000 |
| Neonatal respiratory | 12.5% | 1 | 8 | 0% | 0 | 7 | p = 1.0000 |
| Hip dislocation | 8.3% | 1 | 12 | 42.9% | 3 | 7 | p = 0.1174 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cotta, A.; Souza, L.S.; Carvalho, E.; Feitosa, L.N.; Cunha, A., Jr.; Navarro, M.M.; Valicek, J.; Menezes, M.M.; Neves, S.V.N.; Xavier-Neto, R.; et al. Central Core Disease: Facial Weakness Differentiating Biallelic from Monoallelic Forms. Genes 2022, 13, 760. https://doi.org/10.3390/genes13050760
Cotta A, Souza LS, Carvalho E, Feitosa LN, Cunha A Jr., Navarro MM, Valicek J, Menezes MM, Neves SVN, Xavier-Neto R, et al. Central Core Disease: Facial Weakness Differentiating Biallelic from Monoallelic Forms. Genes. 2022; 13(5):760. https://doi.org/10.3390/genes13050760
Chicago/Turabian StyleCotta, Ana, Lucas Santos Souza, Elmano Carvalho, Leticia Nogueira Feitosa, Antonio Cunha, Jr., Monica Machado Navarro, Jaquelin Valicek, Miriam Melo Menezes, Simone Vilela Nunes Neves, Rafael Xavier-Neto, and et al. 2022. "Central Core Disease: Facial Weakness Differentiating Biallelic from Monoallelic Forms" Genes 13, no. 5: 760. https://doi.org/10.3390/genes13050760