
Major Depressive Disorder: Existing Hypotheses about Pathophysiological Mechanisms and New Genetic Findings


Abstract
:1. Introduction
2. Diagnosis and MDD Phenotypes
3. Available Treatment Options for MDD
4. Pathophysiological Mechanisms of MDD
4.1. Biogenic Amine Hypothesis
4.2. Receptor Hypothesis
4.3. Endocrine System
4.4. The Cytokine Theory
4.5. Neurotrophic Factor Hypothesis
4.6. Neuroplasticity/Neurogenesis Hypothesis
5. Major Risk Factors of MDD
5.1. Environmental Factors
5.2. Genetic Factors and Heritability
6. Poor Replication of MDD-Associated Genes Identified by Candidate Gene Studies
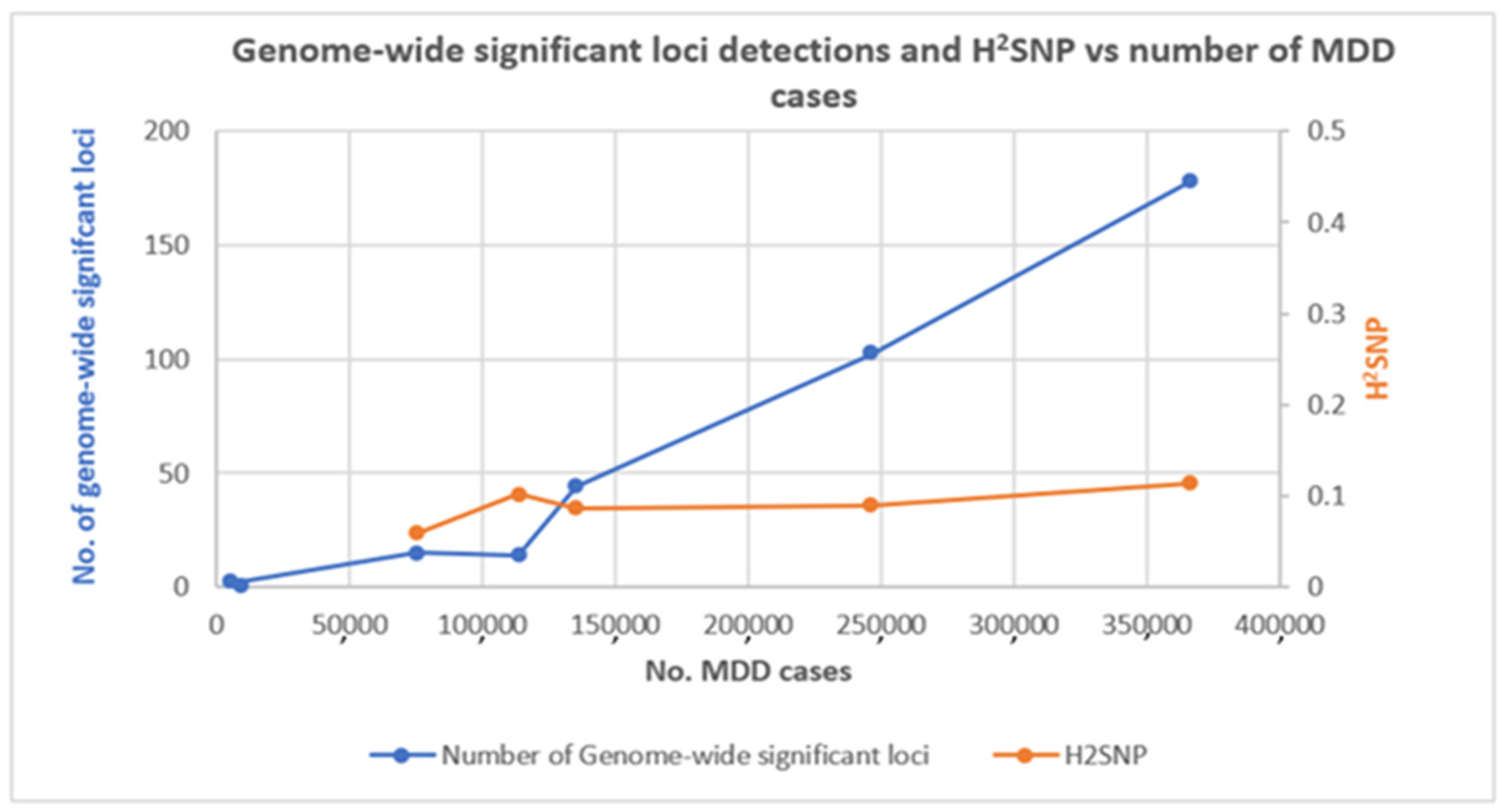
7. Contribution of GWAS to Our Understanding of the Genetic Architecture of MDD
8. GWAS Identify Genetic Correlations between MDD and Other Disorders and Traits
9. Combined Analysis of MDD with Brain-Related Phenotypes Increases Discovery of New Loci
10. Exploring Causal Relationships
11. Rare Variant Analysis of MDD
12. Challenges for Genetic Studies of MDD
13. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kessler, R.C.; Chiu, W.T.; Demler, O.; Merikangas, K.R.; Walters, E.E. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch. Gen. Psychiatry 2005, 62, 617–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Depression and Other Common Mental Disorders: Global Health Estimates; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Bromet, E.; Andrade, L.H.; Hwang, I.; Sampson, N.A.; Alonso, J.; de Girolamo, G.; de Graaf, R.; Demyttenaere, K.; Hu, C.; Iwata, N.; et al. Cross-national epidemiology of DSM-IV major depressive episode. BMC Med. 2011, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Howard, D.M.; Adams, M.J.; Clarke, T.-K.; Hafferty, J.D.; Gibson, J.; Shirali, M.; Coleman, J.R.I.; Hagenaars, S.P.; Ward, J.; Wigmore, E.M.; et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 2019, 22, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wray, N.R.; Ripke, S.; Mattheisen, M.; Trzaskowski, M.; Byrne, E.M.; Abdellaoui, A.; Adams, M.J.; Agerbo, E.; Air, T.M.; Andlauer, T.M.F.; et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 2018, 50, 668–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papakostas, G.I. Major depressive disorder: Psychosocial impairment and key considerations in functional improvement. Am. J. Manag. Care 2009, 15, S316–S321. [Google Scholar]
- Guha, M. Diagnostic and statistical manual of mental disorders: DSM-5. Ref. Rev. 2014, 28, 36–37. [Google Scholar]
- Katon, W.J. Epidemiology and treatment of depression in patients with chronic medical illness. Dialogues Clin. Neurosci. 2011, 13, 7. [Google Scholar] [CrossRef]
- Iannuzzo, R.W.; Jaeger, J.; Goldberg, J.F.; Kafantaris, V.; Sublette, M.E. Development and reliability of the HAM-D/MADRS interview: An integrated depression symptom rating scale. Psychiatry Res. 2006, 145, 21–37. [Google Scholar] [CrossRef]
- McCance-Katz, E.F. The National Survey on Drug Use and Health. Substance Abuse and Mental Health Services Administration. 2017. Available online: https://www.samhsa.gov/data/sites/default/files/nsduh-ppt-09-2018 (accessed on 7 May 2019).
- Dubovsky, S.L.; Ghosh, B.M.; Serotte, J.C.; Cranwell, V. Psychotic depression: Diagnosis, differential diagnosis, and treatment. Psychother. Psychosom. 2021, 90, 160–177. [Google Scholar] [CrossRef]
- Zimmerman, M.; Morgan, T.A.; Stanton, K. The severity of psychiatric disorders. World Psychiatry 2018, 17, 258–275. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Howard, D.; Sibille, E.; Leon French, L. Differential and spatial expression meta-analysis of genes identified in genome-wide association studies of depression. Transl. Psychiatry 2021, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, C.; Hagenaars, S.P.; John, C.; Williams, A.T.; Shrine, N.; Moles, L.; Hanscombe, K.B.; Serretti, A.; Shepherd, D.J.; Free, R.C.; et al. Genetic and clinical characteristics of treatment-resistant depression using primary care records in two UK cohorts. Mol. Psychiatry 2021, 26, 3363–3373. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Liu, X.; Wen, J.; Zhang, Y. Major clinical advances of depression: Now and future. In E3S Web Conference; EDP Sciences: Les Ulis, France, 2021; Volume 292, p. 03102. [Google Scholar]
- Li, Z.; Meihua Ruan, M.; Chen, J.; Fang, Y. Major Depressive Disorder: Advances in Neuroscience Research and Translational Applications. Neurosci. Bull. 2021, 37, 863–880. [Google Scholar] [CrossRef]
- Okbay, A.; Baselmans, B.M.L.; De Neve, J.-E.; Turley, P.; Nivard, M.G.; Fontana, M.A.; Meddens, S.F.W.; Linnér, R.K.; Rietveld, C.A.; Derringer, J.; et al. Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nat. Genet. 2016, 48, 624–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, L.T.; Zarate, C.A., Jr. Depression in the primary care setting. N. Engl. J. Med. 2019, 380, 559–568. [Google Scholar] [CrossRef]
- Kolovos, S.; Kleiboer, A.; Cuijpers, P. Effect of psychotherapy for depression on quality of life: Meta-analysis. Br. J. Psychiatry 2016, 209, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Bains, N.; Abdijadid, S. Major Depressive Disorder. StatPearls [Internet]. 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK559078/ (accessed on 5 October 2021).
- Kessler, R.C. The potential of predictive analytics to provide clinical decision support in depression treatment planning. Curr. Opin. Psychiatry 2018, 31, 32–39. [Google Scholar] [CrossRef]
- Ménard, C.; Hodes, G.E.; Russo, S.J. Pathogenesis of depression: Insights from human and rodent studies. Neuroscience 2016, 321, 138–162. [Google Scholar] [CrossRef] [Green Version]
- Hasler, G. Pathophysiology of depression: Do we have any solid evidence of interest to clinicians? World Psychiatry 2010, 9, 155. [Google Scholar] [CrossRef] [Green Version]
- Shadrina, M.; Bondarenko, E.A.; Slominsky, P.A. Genetics factors in major depression disease. Front. Psychiatry 2018, 9, 334. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-Q.; Wang, Z.Z.; Chen, N.-H. The receptor hypothesis and the pathogenesis of depression: Genetic bases and biological correlates. Pharmacol. Res. 2021, 167, 105542. [Google Scholar] [CrossRef]
- Jesulola, E.; Micalos, P.; Baguley, I.J. Understanding the pathophysiology of depression: From monoamines to the neurogenesis hypothesis model-are we there yet? Behav. Brain Res. 2018, 341, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Girotti, M.; Adler, S.M.; Bulin, S.E.; Fucich, E.A.; Paredes, D.; Morilak, D. Prefrontal cortex executive processes affected by stress in health and disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 85, 161–179. [Google Scholar] [CrossRef] [PubMed]
- Brigitta, B. Pathophysiology of depression and mechanisms of treatment. Dialogues Clin. Neurosci. 2002, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Ashe, K.; Kelso, W.; Farrand, S.; Panetta, J.; Fazio, T.; De Jong, C.; Walterfang, M. Psychiatric and cognitive aspects of phenylketonuria: The limitations of diet and promise of new treatments. Front. Psychiatry 2019, 10, 561. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, J.; Guo, W. Emotional roles of mono-aminergic neurotransmitters in major depressive disorder and anxiety disorders. Front. Psychol. 2018, 9, 2201. [Google Scholar] [CrossRef]
- Stockmeier, C.A.; Rajkowska, G. Cellular abnormalities in depression: Evidence from postmortem brain tissue. Dialogues Clin. Neurosci. 2004, 6, 185. [Google Scholar] [CrossRef]
- Zhou, X.; Xiao, Q.; Xie, L.; Yang, F.; Wang, L.; Tu, J. Astrocyte, a promising target for mood disorder interventions. Front. Mol. Neurosci. 2019, 12, 136. [Google Scholar] [CrossRef]
- Greger, I.H.; Watson, J.F.; Cull-Candy, S.G. Structural and functional architecture of AMPA-type glutamate receptors and their auxiliary proteins. Neuron 2017, 94, 713–730. [Google Scholar] [CrossRef]
- Formicola, D.; Aloia, A.; Sampaolo, S.; Farina, O.; Diodato, D.; Griffiths, L.R.; Gianfrancesco, F.; Di Iorio, D.; Esposito, T. Common variants in the regulative regions of GRIA1 and GRIA3 receptor genes are associated with migraine susceptibility. BMC Med. Genet. 2010, 11, 103. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.H.; Pei, D.; Yang, W.; Cheng, C.; Jeha, S.; Cox, N.J.; Evans, W.E.; Pui, C.-H.; Relling, M.V. Genetic variations in GRIA1 on chromosome 5q33 related to asparaginase hypersensitivity. Clin. Pharmacol. Ther. 2010, 88, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, T.; Arslan, A.; Wisden, W.; Wulff, P. GABAA receptors: Structure and function in the basal ganglia. Prog. Brain Res. 2007, 160, 21–41. [Google Scholar] [PubMed] [Green Version]
- Luscher, B.; Shen, Q.; Sahir, N. The GABAergic deficit hypothesis of major depressive disorder. Mol. Psychiatry 2011, 16, 383–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adell, A. Brain NMDA receptors in schizophrenia and depression. Biomolecules 2020, 10, 947. [Google Scholar] [CrossRef]
- Myers, S.J.; Yuan, H.; Kang, J.-Q.; Tan, F.C.K.; Traynelis, S.F.; Low, C.M. Distinct roles of GRIN2A and GRIN2B variants in neurological conditions. F1000Research 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Shin, W.; Kim, K.; Serraz, B.; Cho, Y.S.; Kim, D.; Kang, M.; Lee, E.-J.; Lee, H.; Bae, Y.C.; Paoletti, P.; et al. Early correction of synaptic long-term depression improves abnormal anxiety-like behavior in adult GluN2B-C456Y-mutant mice. PLoS Biol. 2020, 18, e3000717. [Google Scholar] [CrossRef]
- Aragam, N.; Wang, K.-S.; Anderson, J.L.; Liu, X. TMPRSS9 and GRIN2B are associated with neuroticism: A genome-wide association study in a European sample. J. Mol. Neurosci. 2013, 50, 250–256. [Google Scholar] [CrossRef]
- Yohn, C.N.; Gergues, M.M.; Samuels, B.A. The role of 5-HT receptors in depression. Mol. Brain 2017, 10, 28. [Google Scholar] [CrossRef]
- Lam, D.; Ancelin, M.-L.; Ritchie, K.; Freak-Poli, R.; Saffery, R.; Ryan, J. Genotype-dependent associations between serotonin transporter gene (SLC6A4) DNA methylation and late-life depression. BMC Psychiatry 2018, 18, 282. [Google Scholar] [CrossRef] [Green Version]
- Saavedra, K.; Molina-Márquez, A.M.; Saavedra, N.; Zambrano, T.; Salazar, L.A. Epigenetic modifications of major depressive disorder. Int. J. Mol. Sci. 2016, 17, 1279. [Google Scholar] [CrossRef]
- Murphy, D.L.; Moya, P.R. Human serotonin transporter gene (SLC6A4) variants: Their contributions to understanding pharmacogenomic and other functional G × G and G × E differences in health and disease. Curr. Opin. Pharmacol. 2011, 11, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-León, S.; Janssens, A.C.J.M.; González-Zuloeta Ladd, A.M.; Del-Favero, J.; Claes, S.J.; Oostra, B.A.; van Duijn, C.M. Meta-analyses of genetic studies on major depressive disorder. Mol. Psychiatry 2008, 13, 772–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koning, A.-S.C.; Buurstede, J.C.; van Weert, L.T.C.M.; Meijer, O.C. Glucocorticoid and mineralocorticoid receptors in the brain: A transcriptional perspective. J. Endocr. Soc. 2019, 3, 1917–1930. [Google Scholar] [CrossRef] [PubMed]
- Odaka, H.; Adachi, N.; Numakawa, T. Impact of glucocorticoid on neurogenesis. Neural Regen. Res. 2017, 12, 1028. [Google Scholar]
- Wiley, J.W.; Higgins, G.A.; Athey, B.D. Stress and glucocorticoid receptor transcriptional programming in time and space: Implications for the brain–gut axis. Neurogastroenterol. Motil. 2016, 28, 12–25. [Google Scholar] [CrossRef]
- Menke, A.; Binder, E.B. Epigenetic alterations in depression and antidepressant treatment. Dialogues Clin. Neurosci. 2014, 16, 395. [Google Scholar] [CrossRef]
- Gurevich, E.V.; Gainetdinov, R.R.; Gurevich, V.V. G protein-coupled receptor kinases as regulators of dopamine receptor functions. Pharmacol. Res. 2016, 111, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Saeedi, H.; Remington, G.; Christensen, B.K. Impact of haloperidol, a dopamine D2 antagonist, on cognition and mood. Schizophr. Res. 2006, 85, 222–231. [Google Scholar] [CrossRef]
- Hayden, E.P.; Klein, D.N.; Dougherty, L.R.; Olino, T.M.; Laptook, R.S.; Dyson, M.W.; Bufferd, S.J.; Durbin, C.E.; Sheikh, H.I.; Singh, S.M. The dopamine D2 receptor gene and depressive and anxious symptoms in childhood: Associations and evidence for gene–environment correlation and gene–environment interaction. Psychiatr. Genet. 2010, 20, 304. [Google Scholar] [CrossRef] [Green Version]
- Rajkumar, R.P. Gut hormones as potential therapeutic targets or biomarkers of response in depression: The case of Motilin. Life 2021, 11, 892. [Google Scholar] [CrossRef]
- Scantamburlo, G.; Hansenne, M.; Fuchs, S.; Pitchot, W.; Pinto, E.; Reggers, J.; Ansseau, M.; Legros, J.J. AVP-and OT-neurophysins response to apomorphine and clonidine in major depression. Psychoneuroendocrinology 2005, 30, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Shahid, M.A.; Ashraf, M.A.; Sharma, S. Physiology, thyroid hormone. In StatPearls [Internet]; Treasure Island (FL), StatPearls Publishing: Bethesda, MD, USA, 2018. Available online: https://www.ncbi.nlm.nih.gov/books/NBK500006/ (accessed on 11 October 2021).
- Hage, M.P.; Azar, S.T. The link between thyroid function and depression. J. Thyroid. Res. 2012, 2012, 590648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucchi, R.; Accorroni, A.; Chiellini, G. Update on 3-iodothyronamine and its neurological and metabolic actions. Front. Physiol. 2014, 5, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varghese, F.P.; Brown, E.S. The hypothalamic-pituitary-adrenal axis in major depressive disorder: A brief primer for primary care physicians. J. Clin. Psychiatry 2001, 3, 151. [Google Scholar] [CrossRef]
- Allen, M.J.; Sharma, S. Physiology, adrenocorticotropic hormone (ACTH). In StatPearls [Internet]; Treasure Island (FL), StatPearls Publishing: Bethesda, MD, USA, 2018. Available online: https://www.ncbi.nlm.nih.gov/books/NBK500031/ (accessed on 11 October 2021).
- Leonard, B.E. The concept of depression as a dysfunction of the immune system. In Depression: From Psychopathology to Pharmacotherapy; Karger Publishers: Basel, Switzerland, 2010; pp. 53–71. [Google Scholar]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.-M.; An, J. Cytokines, inflammation and pain. Int. Anesthesiol. Clin. 2007, 45, 27. [Google Scholar] [CrossRef] [Green Version]
- Sproston, N.R.; Ashworth, J.J. Role of C-reactive protein at sites of inflammation and infection. Front. Immunol. 2018, 9, 754. [Google Scholar] [CrossRef]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and its discontents: The role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef] [Green Version]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Maes, M.; Berk, M.; Goehler, L.; Song, C.; Anderson, G.; Gałecki, P.; Leonard, B. Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Med. 2012, 10, 66. [Google Scholar] [CrossRef] [Green Version]
- Amidfar, M.; Réus, G.Z.; de Moura, A.B.; Quevedo, J.; Kim, Y.-K. The role of neurotrophic factors in pathophysiology of major depressive disorder. In Major Depressive Disorder; Springer: Berlin/Heidelberg, Germany, 2021; pp. 257–272. [Google Scholar]
- Eliwa, H.; Brizard, B.; Le Guisquet, A.-M.; Hen, R.; Belzung, C.; Surget, A. Adult neurogenesis augmentation attenuates anhedonia and HPA axis dysregulation in a mouse model of chronic stress and depression. Psychoneuroendocrinology 2021, 124, 105097. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, A.P.; Nikkheslat, N.; Hastings, C.; Nettis, M.A.; Kose, M.; Worrell, C.; Zajkowska, Z.; Mariani, N.; Enache, D.; Lombardo, G.; et al. The influence of comorbid depression and overweight status on peripheral inflammation and cortisol levels. Psychol. Med. 2021, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.J.; Huang, X.F.; Newell, K.A. The kynurenine pathway in major depression: What we know and where to next. Neurosci. Biobehav. Rev. 2021, 127, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Maffioletti, E.; Silva, R.C.; Bortolomasi, M.; Baune, B.T.; Gennarelli, M.; Minelli, A. Molecular biomarkers of electroconvulsive therapy effects and clinical response: Understanding the present to shape the future. Brain Sci. 2021, 11, 1120. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Deyama, S.; Fogaça, M.V. Role of BDNF in the pathophysiology and treatment of depression: Activity-dependent effects distinguish rapid-acting antidepressants. Eur. J. Neurosci. 2021, 53, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Pariante, C.M.; Miller, A.H. Glucocorticoid receptors in major depression: Relevance to pathophysiology and treatment. Biol. Psychiatry 2001, 49, 391–404. [Google Scholar] [CrossRef]
- Walkery, A.; Leader, L.D.; Cooke, E.; VandenBerg, A. Review of allopregnanolone agonist therapy for the treatment of depressive disorders. Drug Des. Dev. Ther. 2021, 15, 3017–3026. [Google Scholar] [CrossRef]
- Chen, S.; Gao, L.; Li, X.; Ye, Y. Allopregnanolone in mood disorders: Mechanism and therapeutic development. Pharmacol. Res. 2021, 169, 105682. [Google Scholar] [CrossRef]
- Bauer, M.; Whybrow, P. Role of thyroid hormone therapy in depressive disorders. J. Endocrinol. Investig. 2021, 44, 2341–2347. [Google Scholar] [CrossRef]
- Urbán, N.; Guillemot, F. Neurogenesis in the embryonic and adult brain: Same regulators, different roles. Front. Cell. Neurosci. 2014, 8, 396. [Google Scholar] [CrossRef] [Green Version]
- Mariani, N.; Cattane, N.; Pariante, C.; Cattaneo, A. Gene expression studies in Depression development and treatment: An overview of the underlying molecular mechanisms and biological processes to identify biomarkers. Transl. Psychiatry 2021, 11, 354. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, A.; Cattane, N.; Begni, V.; Pariante, C.M.; Riva, M.A. The human BDNF gene: Peripheral gene expression and protein levels as biomarkers for psychiatric disorders. Transl. Psychiatry 2016, 6, e958. [Google Scholar] [CrossRef] [PubMed]
- Castrén, E.; Monteggia, L.M. Brain-derived neurotrophic factor signaling in depression and antidepressant action. Biol. Psychiatry 2021, 90, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Nie, Z.; Shu, H.; Kuang, Y.; Chen, X.; Cheng, J.; Yu, S.; Liu, H. The role of BDNF on neural plasticity in depression. Front. Cell. Neurosci. 2020, 14, 82. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.; Alkan, E.; Bhangoo, J.K.; Tenenbaum, H.; Ng-Knight, T. Effects of the COVID-19 lockdown on mental health, wellbeing, sleep, and alcohol use in a UK student sample. Psychiatry Res. 2021, 298, 113819. [Google Scholar] [CrossRef] [PubMed]
- Zunszain, P.A.; Hepgul, N.; Pariante, C.M. Inflammation and depression. In Behavioral Neurobiology of Depression and Its Treatment; Springer: Berlin/Heidelberg, Germany, 2012; pp. 135–151. [Google Scholar]
- Fallah, H.; Rademaker, M. Isotretinoin for acne vulgaris–an update on adverse effects and laboratory monitoring. J. Dermatol. Treat. 2021, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Soyka, M. Rimonabant and depression. Pharmacopsychiatry 2008, 41, 204–205. [Google Scholar] [CrossRef]
- Gupta, M.; Neavin, D.; Liu, D.; Biernacka, J.; Hall-Flavin, D.; Bobo, W.V.; Frye, M.A.; Skime, M.; Jenkins, G.D.; Batzler, A.; et al. TSPAN5, ERICH3 and selective serotonin reuptake inhibitors in major depressive disorder: Pharmacometabolomics-informed pharmacogenomics. Mol. Psychiatry 2016, 21, 1717–1725. [Google Scholar] [CrossRef]
- Wray, N.; Visscher, P. Estimating trait heritability. Nat. Educ. 2008, 1, 29. [Google Scholar]
- van Calker, D.; Serchov, T. The “missing heritability”—Problem in psychiatry: Is the interaction of genetics, epigenetics and transposable elements a potential solution? Neurosci. Biobehav. Rev. 2021, 126, 23–42. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wray, N.R.; Gottesman, I.I. Using summary data from the danish national registers to estimate heritabilities for schizophrenia, bipolar disorder, and major depressive disorder. Front. Genet. 2012, 3, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polubriaginof, F.C.; Vanguri, R.; Quinnies, K.; Belbin, G.M.; Yahi, A.; Salmasian, H.; Lorberbaum, T.; Nwankwo, V.; Li, L.; Shervey, M.M.; et al. Disease heritability inferred from familial relationships reported in medical records. Cell 2018, 173, 1692–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendler, K.S.; Ohlsson, H.; Lichtenstein, P.; Sundquist, J.; Sundquist, K. The genetic epidemiology of treated major depression in Sweden. Am. J. Psychiatry 2018, 175, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Cai, N.; Bigdeli, T.; Kretzschmar, W.W.; Li, Y.; Liang, J.; Song, L.; Hu, J.; Li, Q.; Jin, W.; Hu, Z.; et al. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 2015, 523, 588–591. [Google Scholar]
- Kendall, K.; Van Assche, E.; Andlauer, T.F.M.; Choi, K.W.; Luykx, J.J.; Schulte, E.C.; Lu, Y. The genetic basis of major depression. Psychol. Med. 2021, 51, 2217–2230. [Google Scholar] [CrossRef]
- McIntosh, A.M.; Sullivan, P.F.; Lewis, C.M. Uncovering the genetic architecture of major depression. Neuron 2019, 102, 91–103. [Google Scholar] [CrossRef]
- Lee, K.W.; Woon, P.S.; Teo, Y.Y.; Sim, K. Genome wide association studies (GWAS) and copy number variation (CNV) studies of the major psychoses: What have we learnt? Neurosci. Biobehav. Rev. 2012, 36, 556–571. [Google Scholar] [CrossRef]
- Uffelmann, E.; Huang, Q.Q.; Munung, N.S.; de Vries, J.; Okada, Y.; Martin, A.R.; Martin, H.C.; Lappalainen, T.; Posthuma, D. Genome-wide association studies. Nat. Rev. Methods Primers 2021, 1, 1–21. [Google Scholar] [CrossRef]
- Levey, D.F.; Stein, M.B.; Wendt, F.R.; Pathak, G.A.; Zhou, H.; Aslan, M.; Quaden, R.; Harrington, K.M.; Nuñez, Y.Z.; Overstreet, C.; et al. Bi-ancestral depression GWAS in the Million Veteran Program and meta-analysis in >1.2 million individuals highlight new therapeutic directions. Nat. Neurosci. 2021, 24, 954–963. [Google Scholar] [CrossRef]
- Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium. A mega-analysis of genome-wide association studies for major depressive disorder. Mol. Psychiatry 2013, 18, 497–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hek, K.; Demirkan, A.; Lahti, J.; Terracciano, A.; Teumer, A.; Cornelis, M.C.; Amin, N.; Bakshis, E.; Baumert, J.; Ding, J.; et al. A genome-wide association study of depressive symptoms. Biol. Psychiatry 2013, 73, 667–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyde, C.L.; Nagle, M.W.; Tian, C.; Chen, X.; Paciga, S.A.; Wendland, J.R.; Tung, J.Y.; Hinds, D.A.; Perlis, R.H.; Winslow, A.R. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat. Genet. 2016, 48, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Howard, D.M.; Adams, M.J.; Shirali, M.; Clarke, T.-K.; Marioni, R.E.; Davies, G.; Coleman, J.R.I.; Alloza, C.; Shen, X.; Barbu, M.C.; et al. Genome-wide association study of depression phenotypes in UK Biobank identifies variants in excitatory synaptic pathways. Nat. Commun. 2018, 9, 1470. [Google Scholar] [CrossRef] [Green Version]
- Wani, A.L.; Bhat, S.A.; Ara, A. Omega-3 fatty acids and the treatment of depression: A review of scientific evidence. Integr. Med. Res. 2015, 4, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, P.-H.; Voss, M.; Haug-Kröper, M.; Schröder, B.; Schepers, U.; Bräse, S.; Haass, C.; Lichtenthaler, S.F.; Fluhrer, R. Secretome analysis identifies novel signal peptide peptidase-like 3 (SPPL3) substrates and reveals a role of SPPL3 in multiple golgi glycosylation pathways. Mol. Cell. Proteom. 2015, 14, 1584–1598. [Google Scholar] [CrossRef] [Green Version]
- Xie, P. TRAF molecules in cell signaling and in human diseases. J. Mol. Signal. 2013, 8, 7. [Google Scholar] [CrossRef]
- Cruz, P.M.R.; Cossins, J.; Beeson, D.; Vincent, A. The neuromuscular junction in health and disease: Molecular mechanisms governing synaptic formation and homeostasis. Front. Mol. Neurosci. 2020, 13, 610964. [Google Scholar] [CrossRef]
- Caspi, A.; Moffitt, T.E. All for one and one for all: Mental disorders in one dimension. Am. J. Psychiatry 2018, 175, 831–844. [Google Scholar] [CrossRef] [Green Version]
- Polderman, T.J.C.; Benyamin, B.; de Leeuw, C.A.; Sullivan, P.F.; van Bochoven, A.; Visscher, P.M.; Posthuma, D. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat. Genet. 2015, 47, 702–709. [Google Scholar] [CrossRef] [Green Version]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 years of GWAS discovery: Biology, function, and translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, K.; Stringer, S.; Frei, O.; Mirkov, M.U.; de Leeuw, C.; Polderman, T.J.C.; van der Sluis, S.; Andreassen, O.A.; Neale, B.M.; Posthuma, D. A global overview of pleiotropy and genetic architecture in complex traits. Nat. Genet. 2019, 51, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.R.; Gaspar, H.A.; Bryois, J.; Bipolar Disorder Working Group of the Psychiatric Genomics Consortium; Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium; Breen, G. The genetics of the mood disorder spectrum: Genome-wide association analyses of more than 185,000 cases and 439,000 controls. Biol. Psychiatry 2020, 88, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Ohi, K.; Otowa, T.; Shimada, M.; Sasaki, T.; Tanii, H. Shared genetic etiology between anxiety disorders and psychiatric and related intermediate phenotypes. Psychol. Med. 2020, 50, 692–704. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Gao, Y.; Chen, M.; Zhang, X.; Yue, W.; Zhang, D.; Yu, H. Overlapping common genetic architecture between major depressive disorders and anxiety and stress-related disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2022, 113, 110450. [Google Scholar] [CrossRef]
- Bahrami, S.; Shadrin, A.; Frei, O.; O’Connell, K.S.; Bettella, F.; Krull, F.; Fan, C.C.; Røssberg, J.I.; Hindley, G.; Ueland, T.; et al. Genetic loci shared between major depression and intelligence with mixed directions of effect. Nat. Hum. Behav. 2021, 5, 795–801. [Google Scholar] [CrossRef]
- Lutz, M.W.; Sprague, D.; Barrera, J.; Chiba-Falek, O. Shared genetic etiology underlying Alzheimer’s disease and major depressive disorder. Transl. Psychiatry 2020, 10, 88. [Google Scholar] [CrossRef] [Green Version]
- Powell, V.; Martin, J.; Thapar, A.; Rice, F.; Anney, R.J.L. Investigating regions of shared genetic variation in attention deficit/hyperactivity disorder and major depressive disorder: A GWAS meta-analysis. Sci. Rep. 2021, 11, 7353. [Google Scholar] [CrossRef]
- Baranova, A.; Cao, H.; Zhang, F. Shared genetic liability and causal effects between major depressive disorder and insomnia. Hum. Mol. Genet. 2021, 30, 2053–2214. [Google Scholar] [CrossRef]
- Davey Smith, G.; Hemani, G. Mendelian randomization: Genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 2014, 23, R89–R98. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.; Meng, W.; Hägg, S.; Burgess, S.; Jiang, X. Reciprocal interaction between depression and pain: Results from a comprehensive bidirectional Mendelian randomization study and functional annotation analysis. Pain 2022, 163, e40–e48. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, J.; Yin, Z.; Wang, L.; Peng, L. The association between depression and metabolic syndrome and its components: A bidirectional two-sample Mendelian randomization study. Transl. Psychiatry 2021, 11, 633. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Cao, H.; Baranova, A. Shared genetic liability and causal associations between major depressive disorder and cardiovascular diseases. Front. Cardiovasc. Med. 2021, 8, 735136. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Xu, Z.; Noordam, R.; van Heemst, D.; Li-Gao, R. Depression and inflammatory bowel disease: A bidirectional two-sample Mendelian randomization study. J. Crohns Colitis 2021, jjab191. [Google Scholar] [CrossRef] [PubMed]
- Barowsky, S.; Jung, J.-Y.; Nesbit, N.; Silberstein, M.; Fava, M.; Loggia, M.L.; Smoller, J.W.; Lee, P.H. Cross-disorder genomics data analysis elucidates a shared genetic basis between major depression and osteoarthritis pain. Front. Genet. 2021, 12, 687687. [Google Scholar] [CrossRef]
- Cao, H.; Li, S.; Baranova, B.; Zhang, F. Shared genetic liability between major depressive disorder and atopic diseases. Front. Immunol. 2021, 12, 665160. [Google Scholar] [CrossRef]
- Perry, B.I.; Upthegrovecd, R.; Kappelmannef, N.; Jonesab, P.B.; Burgessg, S.; Khandaker, G.M. Associations of immunological proteins/traits with schizophrenia, major depression and bipolar disorder: A bi-directional two-sample mendelian randomization study. Brain Behav. Immun. 2021, 97, 176–185. [Google Scholar] [CrossRef]
- Kendall, K.M.; Rees, E.; Bracher-Smith, M.; Legge, S.; Riglin, L.; Zammit, S.; O’Donovan, M.C.; Owen, M.J.; Jones, I.; Kirov, G.; et al. Association of rare copy number variants with risk of depression. JAMA Psychiatry 2019, 76, 818–825. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Abdellaoui, A.; Rucker, J.; de Jong, S.; Potash, J.B.; Weissman, M.M.; Shi, J.; Knowles, J.A.; Pato, C.; Pato, M.; et al. Genome-wide burden of rare short deletions is enriched in major depressive disorder in four cohorts. Biol. Psychiatry 2019, 85, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.Y. A simple and accurate method to determine genomewide significance for association tests in sequencing studies. Genet. Epidemiol. 2019, 43, 365–372. [Google Scholar] [CrossRef]
- Kang, H.-J.; Park, Y.; Yoo, K.-H.; Kim, K.-T.; Kim, E.-S.; Kim, J.-W.; Kim, S.-W.; Shin, I.-S.; Yoon, J.-S.; Kim, J.H.; et al. Sex differences in the genetic architecture of depression. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, L.; Jiang, B.; Sun, Y.; Li, M.; Wu, H.; Zhang, N.; Sun, X.; Qin, S. Large-scale whole-exome sequencing association study identifies FOXH1 gene and sphingolipid metabolism pathway influencing major depressive disorder. CNS Neurosci. Ther. 2021, 27, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Curtis, D. Analysis of 200 000 exome-sequenced UK Biobank subjects fails to identify genes influencing probability of developing a mood disorder resulting in psychiatric referral. Psychiatr. Genet. 2021, 31, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Levinson, D.F.; Mostafavi, S.; Milaneschi, Y.; Rivera, M.; Ripke, S.; Wray, N.R.; Sullivan, P.F. Genetic studies of major depressive disorder: Why are there no genome-wide association study findings and what can we do about it? Biol. Psychiatry 2014, 76, 510–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smoller, J.W.; Andreassen, O.A.; Edenberg, H.J.; Faraone, S.V.; Glatt, S.J.; Kendler, K.S. Psychiatric genetics and the structure of psychopathology. Mol. Psychiatry 2019, 24, 409–420. [Google Scholar] [CrossRef]
- Sullivan, P.F.; Neale, M.C.; Kendler, K.S. Genetic epidemiology of major depression: Review and meta-analysis. Am. J. Psychiatry 2000, 157, 1552–1562. [Google Scholar] [CrossRef]
- Young, A.I. Solving the missing heritability problem. PLoS Genet. 2019, 15, e1008222. [Google Scholar] [CrossRef] [Green Version]
- Ripke, S.; Walters, J.T.; O’Donovan, M.C.; Schizophrenia Working Group of the Psychiatric Genomics Consortium. Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia. medRxiv 2020. [Google Scholar] [CrossRef]
- Liebers, D.T.; Pirooznia, M.; Ganna, A.; Goes, F.S.; Bipolar Genome Study (BiGS). Discriminating bipolar depression from major depressive disorder with polygenic risk scores. Psychol. Med. 2021, 51, 1451–1458. [Google Scholar] [CrossRef]
- Ward, J.; Graham, N.; Strawbridge, R.J.; Ferguson, A.; Jenkins, G.; Chen, W.; Hodgson, K.; Frye, M.; Weinshilboum, R.; Uher, R.; et al. Polygenic risk scores for major depressive disorder and neuroticism as predictors of antidepressant response: Meta-analysis of three treatment cohorts. PLoS ONE 2018, 13, e0203896. [Google Scholar] [CrossRef] [Green Version]
- Fullerton, J.M.; Nurnberger, J.I. Polygenic risk scores in psychiatry: Will they be useful for clinicians? F1000Research 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirugo, G.; Williams, S.M.; Tishkoff, S.A. The missing diversity in human genetic studies. Cell 2019, 177, 26–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannakopoulou, O.; Lin, K.; Meng, X.; Su, M.-H.; Kuo, P.-H.; Peterson, R.E.; Awasthi, S.; Moscati, A.; Coleman, J.R.I.; Bass, N.; et al. The genetic architecture of depression in individuals of East Asian ancestry: A genome-wide association study. JAMA Psychiatry 2021, 78, 1258–1269. [Google Scholar] [CrossRef] [PubMed]
- Peterson, R.E.; Cai, N.; Bigdeli, T.B.; Li, Y.; Reimers, M.; Nikulova, A.; Webb, B.T.; Bacanu, S.-A.; Riley, B.P.; Flint, J.; et al. The genetic architecture of major depressive disorder in han Chinese women. JAMA Psychiatry 2017, 74, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Howard, D.M.; Adams, M.J.; Hill, W.D.; Clarke, T.-K.; Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium; Deary, J.J.; Whalley, H.C.; Mclntosh, A.M. A phenome-wide association and Mendelian randomisation study of polygenic risk for depression in UK Biobank. Nat. Commun. 2020, 11, 2301. [Google Scholar] [CrossRef]


{kind=link}
| Pharmacological Class | Drugs |
|---|---|
| Selective seretonin reuptake inhibitors (SSRIs) | Citalopram, escitalopram, fluoxetine, fluvoxamine, sertraline and paroxetine |
| Serotonin and norepinephrine reuptake in-hibitors (SNRIs) | Duloxetine, desvenlafaxine and venlafaxine |
| Monoamine oxidase inhibitors (MAOIs) | Isocarboxazid, phenelzine, selegiline and tranylcypromine |
| Tricyclic antidepressants (TCAs) | Amitriptyline, desipramine, doxepine, maprotiline, nortriptyline, protriptyline and imipramine |
| Noradrenergic and specific serotonergic modulators | Mirtazapine |
| Norepinephrine-dopamine reuptake inhibitors | Bupropion |
| Multimodal antidepressants | Vortioxetine |
| Serotonin modulators | Trazodone and nefazodone |
| MT1/MT2 agonists and 5-HT2C antagonists | Agomelatine |
| Serotonin reuptake inhibitors and 5-HT1A-receptor partial agonists | Vilazodone |
| Neurosteroids | Bresanolone |
| Newer agents like NMDA receptor antagonists | Esketamine |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamran, M.; Bibi, F.; ur. Rehman, A.; Morris, D.W. Major Depressive Disorder: Existing Hypotheses about Pathophysiological Mechanisms and New Genetic Findings. Genes 2022, 13, 646. https://doi.org/10.3390/genes13040646
Kamran M, Bibi F, ur. Rehman A, Morris DW. Major Depressive Disorder: Existing Hypotheses about Pathophysiological Mechanisms and New Genetic Findings. Genes. 2022; 13(4):646. https://doi.org/10.3390/genes13040646
Chicago/Turabian StyleKamran, Muhammad, Farhana Bibi, Asim. ur. Rehman, and Derek W. Morris. 2022. "Major Depressive Disorder: Existing Hypotheses about Pathophysiological Mechanisms and New Genetic Findings" Genes 13, no. 4: 646. https://doi.org/10.3390/genes13040646