
Novel Protein–Protein Interactions Highlighting the Crosstalk between Hypoplastic Left Heart Syndrome, Ciliopathies and Neurodevelopmental Delays

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Compilation of HLHS-Associated Genes and Prediction of Novel Interactions
2.2. Identification of Network Modules
2.3. Functional Enrichment Analysis
2.4. Gene Expression Enrichment Analysis
2.5. Network Overlap Analysis
3. Results
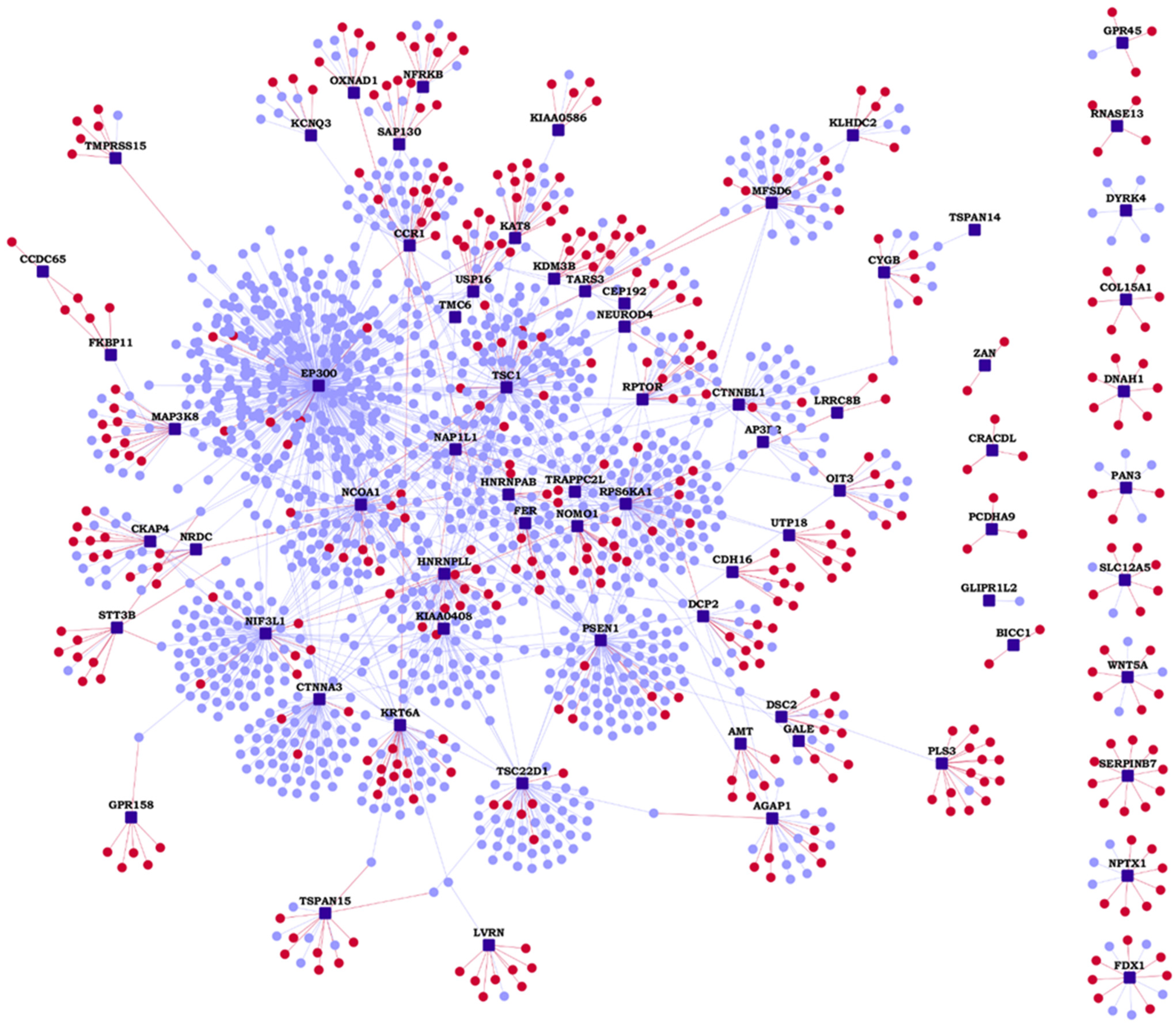
3.1. Wiki-HLHS: A Webserver of HLHS PPIs
3.2. Identification of Network Modules from the HLHS Interactome
3.3. Functional Enrichment for Human Diseases in the HLHS Interactome
3.4. GO Biological Process Enrichment and Overlap with HLHS Transcriptome Datasets
3.5. HLHS and Developmental Delay
3.6. HLHS and Microcephaly
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zaidi, S.; Brueckner, M. Genetics and genomics of congenital heart disease. Circ. Res. 2017, 120, 923–940. [Google Scholar] [CrossRef] [PubMed]
- Gobergs, R.; Salputra, E.; Lubaua, I. Hypoplastic left heart syndrome: A review. Acta Med. Litu. 2016, 23, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Šamánek, M.; Slavík, Z.; Zbořilová, B.; Hroboňová, V.; Voříšková, M.; Skovranek, J. Prevalence, treatment, and outcome of heart disease in live-born children: A prospective analysis of 91,823 live-born children. Pediatr. Cardiol. 1989, 10, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Hamzah, M.; Othman, H.F.; Baloglu, O.; Aly, H. Outcomes of hypoplastic left heart syndrome: Analysis of National Inpatient Sample Database 1998–2004 versus 2005–2014. Eur. J. Pediatr. 2019, 179, 309–316. [Google Scholar] [CrossRef] [PubMed]
- D’Udekem, Y.; Iyengar, A.J.; Galati, J.C.; Forsdick, V.; Weintraub, R.G.; Wheaton, G.R.; Bullock, A.; Justo, R.N.; Grigg, L.E.; Sholler, G.F. Redefining expectations of long-term survival after the Fontan procedure: Twenty-five years of follow-up from the entire population of Australia and New Zealand. Circulation 2014, 130, S32–S38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsoufi, B.; Mori, M.; Gillespie, S.; Schlosser, B.; Slesnick, T.; Kogon, B.; Kim, D.; Sachdeva, R.; Kanter, K. Impact of patient characteristics and anatomy on results of norwood operation for hypoplastic left heart syndrome. Ann. Thorac. Surg. 2015, 100, 591–598. [Google Scholar] [CrossRef] [Green Version]
- Siffel, C.; Riehle-Colarusso, T.; Oster, M.E.; Correa, A. Survival of children with hypoplastic left heart syndrome. Pediatrics 2015, 136, e864–e870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, B.S.; Lipkin, P.H.; Newburger, J.W.; Peacock, G.; Gerdes, M.; Gaynor, J.W.; Mussatto, K.A.; Uzark, K.; Goldberg, C.S.; Johnson, W.H., Jr. Neurodevelopmental outcomes in children with congenital heart disease: Evaluation and management: A scientific statement from the American Heart Association. Circulation 2012, 126, 1143–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinton, R.B.; Andelfinger, G.; Sekar, P.; Hinton, A.C.; Gendron, R.L.; Michelfelder, E.C.; Robitaille, Y.; Benson, D.W. Prenatal head growth and white matter injury in hypoplastic left heart syndrome. Pediatr. Res. 2008, 64, 364–369. [Google Scholar] [CrossRef] [Green Version]
- Hinton, R.B.; Martin, L.J.; Tabangin, M.E.; Mazwi, M.L.; Cripe, L.H.; Benson, D.W. Hypoplastic left heart syndrome is heritable. J. Am. Coll. Cardiol. 2007, 50, 1590–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, K.; Pignatelli, R.; Lewin, M.; Ho, T.; Fernbach, S.; Menesses, A.; Lam, W.; Leal, S.M.; Kaplan, N.; Schliekelman, P.; et al. Inheritance analysis of congenital left ventricular outflow tract obstruction malformations: Segregation, multiplex relative risk, and heritability. Am. J. Med. Genet. Part A 2005, 134A, 180–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Yagi, H.; Saeed, S.; Bais, A.S.; Gabriel, G.C.; Chen, Z.; Peterson, K.A.; Li, Y.; Schwartz, M.C.; Reynolds, W.T.; et al. The complex genetics of hypoplastic left heart syndrome. Nat. Genet. 2017, 49, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- McBride, K.L.; Zender, G.A.; Fitzgerald-Butt, S.M.; Koehler, D.; Menesses-Diaz, A.; Fernbach, S.; Lee, K.; Towbin, J.A.; Leal, S.; Belmont, J. Linkage analysis of left ventricular outflow tract malformations (aortic valve stenosis, coarctation of the aorta, and hypoplastic left heart syndrome). Eur. J. Hum. Genet. 2009, 17, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.; Choi, M.; Wakimoto, H.; Ma, L.; Jiang, J.; Overton, J.D.; Romano-Adesman, A.; Bjornson, R.D.; Breitbart, R.E.; Brown, K.K.; et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013, 498, 220–223. [Google Scholar] [CrossRef] [Green Version]
- Theis, J.L.; Hu, J.J.; Sundsbak, R.S.; Evans, J.M.; Bamlet, W.R.; Qureshi, M.Y.; O’Leary, P.W.; Olson, T.M. Genetic Association Between Hypoplastic Left Heart Syndrome and Cardiomyopathies. Circ. Genom. Precis. Med. 2021, 14, e003126. [Google Scholar] [CrossRef]
- Reuter, M.S.; Chaturvedi, R.R.; Liston, E.; Manshaei, R.; Aul, R.B.; Bowdin, S.; Cohn, I.; Curtis, M.; Dhir, P.; Hayeems, R.Z.; et al. The Cardiac Genome Clinic: Implementing genome sequencing in pediatric heart disease. Genet. Med. 2020, 22, 1015–1024. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.K.; Deshmukh, V.; Nutter, C.A.; Jaworski, E.; Jin, W.; Wadhwa, L.; Abata, J.; Ricci, M.; Lincoln, J.; Martin, J.F.; et al. Rbfox2 function in RNA metabolism is impaired in hypoplastic left heart syndrome patient hearts. Sci. Rep. 2016, 6, 30896. [Google Scholar] [CrossRef]
- Gill, H.K.; Parsons, S.R.; Spalluto, C.; Davies, A.F.; Knorz, V.J.; Burlinson, C.E.; Ng, B.L.; Carter, N.P.; Ogilvie, C.M.; Wilson, D.I.; et al. Separation of the PROX1 gene from upstream conserved elements in a complex inversion/translocation patient with hypoplastic left heart. Eur. J. Hum. Genet. 2009, 17, 1423–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theis, J.L.; Vogler, G.; Missinato, M.A.; Li, X.; Nielsen, T.; Zeng, X.-X.I.; Martinez-Fernandez, A.; Walls, S.M.; Kervadec, A.; Kezos, J.N.; et al. Patient-specific genomics and cross-species functional analysis implicate LRP2 in hypoplastic left heart syndrome. eLife 2020, 9, e59554. [Google Scholar] [CrossRef] [PubMed]
- Homsy, J.; Zaidi, S.; Shen, Y.; Ware, J.S.; Samocha, K.E.; Karczewski, K.J.; DePalma, S.R.; McKean, D.; Wakimoto, H.; Gorham, J.; et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 2015, 350, 1262–1266. [Google Scholar] [CrossRef] [Green Version]
- Ganapathiraju, M.K.; Thahir, M.; Handen, A.; Sarkar, S.N.; Sweet, R.A.; Nimgaonkar, V.L.; Loscher, C.E.; Bauer, E.M.; Chaparala, S. Schizophrenia interactome with 504 novel protein–protein interactions. NPJ Schizophr. 2016, 2, 16012. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Hao, T.; Shaw, C.; Patel, A.J.; Szabó, G.; Rual, J.-F.; Fisk, C.J.; Li, N.; Smolyar, A.; Hill, D.E.; et al. A Protein–protein interaction network for human inherited ataxias and disorders of purkinje cell degeneration. Cell 2006, 125, 801–814. [Google Scholar] [CrossRef] [Green Version]
- Sakai, Y.; Shaw, C.A.; Dawson, B.C.; Dugas, D.V.; Al-Mohtaseb, Z.; Hill, D.E.; Zoghbi, H.Y. Protein interactome reveals converging molecular pathways among autism disorders. Sci. Transl. Med. 2011, 3, 86ra49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, T.S.K.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human protein reference database-2009 update. Nucleic Acids Res. 2008, 37, D767–D772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, C.; Breitkreutz, B.-J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Zhang, Y.; Ghosh, A.; Cuevas, R.A.; Forero, A.; Dhar, J.; Ibsen, M.S.; Schmid-Burgk, J.L.; Schmidt, T.; Ganapathiraju, M.; et al. Antiviral activity of human oasl protein is mediated by enhancing signaling of the RIG-I RNA Sensor. Immunity 2014, 40, 936–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karunakaran, K.B.; Yanamala, N.; Boyce, G.; Becich, M.J.; Ganapathiraju, M.K. Malignant pleural mesothelioma interactome with 364 novel protein-protein interactions. Cancers 2021, 13, 1660. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Klena, N.T.; Gabriel, G.C.; Liu, X.; Kim, A.J.; Lemke, K.; Chen, Y.; Chatterjee, B.; Devine, W.; Damerla, R.R.; et al. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature 2015, 521, 520–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Demir, E.; Schultz, N.; Taylor, B.S.; Sander, C. Automated network analysis identifies core pathways in glioblastoma. PLoS ONE 2010, 5, e8918. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, J. In search of the biological significance of modular structures in protein networks. PLoS Comput. Biol. 2007, 3, e107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.O. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croft, D.; Mundo, A.F.; Haw, R.; Orlic-Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.V.; Gillespie, M.; Kamdar, M.R.; et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2013, 42, D472–D477. [Google Scholar] [CrossRef] [PubMed]
- Hamosh, A.; Scott, A.F.; Amberger, J.S.; Bocchini, C.A.; Valle, D.; McKusick, V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2002, 30, 52–55. [Google Scholar] [CrossRef]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2016, 45, gkw943. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [Green Version]
- Consortium, G. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Tuteja, G. TissueEnrich: Tissue-specific gene enrichment analysis. Bioinformatics 2019, 35, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Stamatoyannopoulos, J.A.; Snyder, M.; Hardison, R.; Ren, B.; Gingeras, T.; Gilbert, D.M.; Groudine, M.; Bender, M.; Kaul, R.; Canfield, T. An encyclopedia of mouse DNA elements (Mouse ENCODE). Genome Biol. 2012, 13, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Dunham, B.; Ganapathiraju, M.K. Benchmark Evaluation of Protein–Protein Interaction Prediction Algorithms. Molecules 2020, 27, 41. [Google Scholar] [CrossRef]
- Gaber, N.; Gagliardi, M.; Patel, P.; Kinnear, C.; Zhang, C.; Chitayat, D.; Shannon, P.; Jaeggi, E.; Tabori, U.; Keller, G.; et al. Fetal Reprogramming and Senescence in Hypoplastic Left Heart Syndrome and in Human Pluripotent Stem Cells during Cardiac Differentiation. Am. J. Pathol. 2013, 183, 720–734. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Jin, K.; Bais, A.S.; Zhu, W.; Yagi, H.; Feinstein, T.N.; Nguyen, P.; Criscione, J.; Liu, X.; Beutner, G.; et al. Uncompensated mitochondrial mediated oxidative stress underlies heart failure in an iPSC-derived model of congenital heart disease. Cell Stem Cell, 2022; in press. [Google Scholar] [CrossRef]
- Becerra, J.E.; Khoury, M.J.; Cordero, J.F.; Erickson, J.D. Diabetes mellitus during pregnancy and the risks for specific birth defects: A population-based case-control study. Pediatrics 1990, 85, 1–9. [Google Scholar] [CrossRef]
- Bagge, C.N.; Henderson, V.W.; Laursen, H.B.; Adelborg, K.; Olsen, M.; Madsen, N.L. Risk of dementia in adults with congenital heart disease: Population-based cohort study. Circulation 2018, 137, 1912–1920. [Google Scholar] [CrossRef]
- Komatsu, H.; Inui, A.; Kishiki, K.; Kawai, H.; Yoshio, S.; Osawa, Y.; Kanto, T.; Fujisawa, T. Liver disease secondary to congenital heart disease in children. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Kogiso, T.; Tokushige, K. Fontan-associated liver disease and hepatocellular carcinoma in adults. Sci. Rep. 2020, 10, 21742. [Google Scholar] [CrossRef]
- Ricci, M.; Xu, Y.; Hammond, H.L.; Willoughby, D.A.; Nathanson, L.; Rodríguez, M.M.; Vatta, M.; Lipshultz, S.E.; Lincoln, J. Myocardial alternative RNA splicing and gene expression profiling in early stage hypoplastic left heart syndrome. PLoS ONE 2012, 7, e29784. [Google Scholar] [CrossRef]
- Yang, C.; Xu, Y.; Yu, M.; Lee, D.; Alharti, S.; Hellen, N.; Shaik, N.A.; Banaganapalli, B.; Mohamoud, H.S.A.; Elango, R.; et al. Induced pluripotent stem cell modelling of HLHS underlines the contribution of dysfunctional NOTCH signalling to impaired cardiogenesis. Hum. Mol. Genet. 2017, 26, 3031–3045. [Google Scholar] [CrossRef] [PubMed]
- Carcamo-Orive, I.; Hoffman, G.E.; Cundiff, P.; Beckmann, N.D.; D’Souza, S.L.; Knowles, J.W.; Patel, A.; Papatsenko, D.; Abbasi, F.; Reaven, G.M.; et al. Analysis of transcriptional variability in a large human ipsc library reveals genetic and non-genetic determinants of heterogeneity. Cell Stem Cell 2016, 20, 518–532.e519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagi, H.; Liu, X.; Gabriel, G.C.; Wu, Y.; Peterson, K.; Murray, S.A.; Aronow, B.J.; Martin, L.J.; Benson, D.W.; Lo, C.W. The Genetic Landscape of Hypoplastic Left Heart Syndrome. Pediatr. Cardiol. 2018, 39, 1069–1081. [Google Scholar] [CrossRef] [PubMed]
- Breschi, A.; Gingeras, T.R.; Guigó, A.B.R. Comparative transcriptomics in human and mouse. Nat. Rev. Genet. 2017, 18, 425–440. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Karunakaran, K.B.; Chaparala, S.; Lo, C.W.; Ganapathiraju, M.K. Cilia interactome with predicted protein–protein interactions reveals connections to Alzheimer’s disease, aging and other neuropsychiatric processes. Sci. Rep. 2020, 10, 15629. [Google Scholar]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef]
- Geddes, G.C.; Stamm, K.; Mitchell, M.; Mussatto, K.A.; Tomita-Mitchell, A. Ciliopathy variant burden and developmental delay in children with hypoplastic left heart syndrome. Genet. Med. 2017, 19, 711–714. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Mantilla, A.J.; Moreno-De-Luca, A.; Ledbetter, D.H.; Martin, C.L. A cross-disorder method to identify novel candidate genes for developmental brain disorders. JAMA Psychiatry 2016, 73, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Hangge, P.T.; Cnota, J.F.; Woo, J.G.; Hinton, A.C.; Divanovic, A.A.; Manning, P.B.; Ittenbach, R.F.; Hinton, R.B. Microcephaly is associated with early adverse neurologic outcomes in hypoplastic left heart syndrome. Pediatr. Res. 2013, 74, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Cacheiro, P.; Haendel, M.A.; Smedley, D.; Consortium, I.M.P.; Initiative, M. New models for human disease from the International Mouse Phenotyping Consortium. Mamm. Genome 2019, 30, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2010, 12, 56–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, H.N.; Olbrych, S.K.; Smith, K.L.; Cnota, J.F.; Habli, M.; Ramos-Gonzales, O.; Owens, K.J.; Hinton, A.C.; Polzin, W.J.; Muglia, L.J.; et al. Hypoplastic left heart syndrome is associated with structural and vascular placental abnormalities and leptin dysregulation. Placenta 2015, 36, 1078–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, A.G.; Bolande, R.P. The liver in congenital heart disease. Effects of infantile coarctation of the aorta and the hypoplastic left heart syndrome in infancy. Am. J. Dis. Child. 1970, 119, 390–394. [Google Scholar] [CrossRef]
- Monzen, K.; Hiroi, Y.; Kudoh, S.; Akazawa, H.; Oka, T.; Takimoto, E.; Hayashi, D.; Hosoda, T.; Kawabata, M.; Miyazono, K.; et al. Smads, Tak1, and Their Common Target Atf-2 Play a Critical Role in Cardiomyocyte Differentiation. J. Cell Biol. 2001, 153, 687–698. [Google Scholar] [CrossRef] [Green Version]
- Yoon, W.H.; Sandoval, H.; Nagarkar-Jaiswal, S.; Jaiswal, M.; Yamamoto, S.; Haelterman, N.A.; Putluri, N.; Putluri, V.; Sreekumar, A.; Tos, T. Loss of nardilysin, a mitochondrial co-chaperone for α-ketoglutarate dehydrogenase, promotes mTORC1 activation and neurodegeneration. Neuron 2017, 93, 115–131. [Google Scholar] [CrossRef] [Green Version]
- Sadakata, T.; Furuichi, T. Identification and mRNA expression of Ogdh, QP-C, and two predicted genes in the postnatal mouse brain. Neurosci. Lett. 2006, 405, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.; Al-Aklabi, M.; Guerra, G.G. Chronic kidney disease in congenital heart disease patients: A narrative review of evidence. Can. J. Kidney Health Dis. 2015, 2, 27. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Chen, L.; Yang, T.; Huang, P.; Wang, L.; Zhao, L.; Zhang, S.; Ye, Z.; Chen, L.; Zheng, Z.; et al. Congenital heart disease and risk of cardiovascular disease: A meta-analysis of cohort studies. J. Am. Heart Assoc. 2019, 8, e012030. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Differentially Expressed in Cardiomyocytes from iPSCS of 5 HLHS Patients Versus 2 Controls (GSE92447) | Differentially Expressed in HLHS-Right Ventricle Versus Control-Left Ventricle/Control-Right Ventricle (GSE23959) | Affected by Alternative Splicing in HLHS-Right Ventricle Versus Control-Right Ventricle/Control-Left Ventricle (GSE23959) | Differentially Expressed in 25 Days old iPSC-Derived Cardiomyocytes from 1 HLHS Proband Versus Parents | Total Count |
|---|---|---|---|---|---|
| DBN1 | ✓ | ✓ | ✓ | 3 | |
| MYL9 | ✓ | ✓ | 2 | ||
| ASCC3 | ✓ | ✓ | 2 | ||
| CDH5 | ✓ | ✓ | 2 | ||
| CKB | ✓ | ✓ | 2 | ||
| GART | ✓ | ✓ | 2 | ||
| PWP1 | ✓ | ✓ | 2 | ||
| TFPI2 | ✓ | ✓ | 2 | ||
| THBS1 | ✓ | ✓ | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karunakaran, K.B.; Gabriel, G.C.; Balakrishnan, N.; Lo, C.W.; Ganapathiraju, M.K. Novel Protein–Protein Interactions Highlighting the Crosstalk between Hypoplastic Left Heart Syndrome, Ciliopathies and Neurodevelopmental Delays. Genes 2022, 13, 627. https://doi.org/10.3390/genes13040627
Karunakaran KB, Gabriel GC, Balakrishnan N, Lo CW, Ganapathiraju MK. Novel Protein–Protein Interactions Highlighting the Crosstalk between Hypoplastic Left Heart Syndrome, Ciliopathies and Neurodevelopmental Delays. Genes. 2022; 13(4):627. https://doi.org/10.3390/genes13040627
Chicago/Turabian StyleKarunakaran, Kalyani B., George C. Gabriel, Narayanaswamy Balakrishnan, Cecilia W. Lo, and Madhavi K. Ganapathiraju. 2022. "Novel Protein–Protein Interactions Highlighting the Crosstalk between Hypoplastic Left Heart Syndrome, Ciliopathies and Neurodevelopmental Delays" Genes 13, no. 4: 627. https://doi.org/10.3390/genes13040627