
Using Transcriptome Analysis to Explore Gray Mold Resistance-Related Genes in Onion (Alliumcepa L.)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. RAPD-PCR and Analysis of Genetic Relationships
2.3. RNA Sequencing
2.4. Differential Gene Expression Analysis
2.5. Analysis of Gene Network and Time-Series Expression
2.6. RNA Extraction and Quantitative Real-Time PCR (qRT-PCR) Analysis
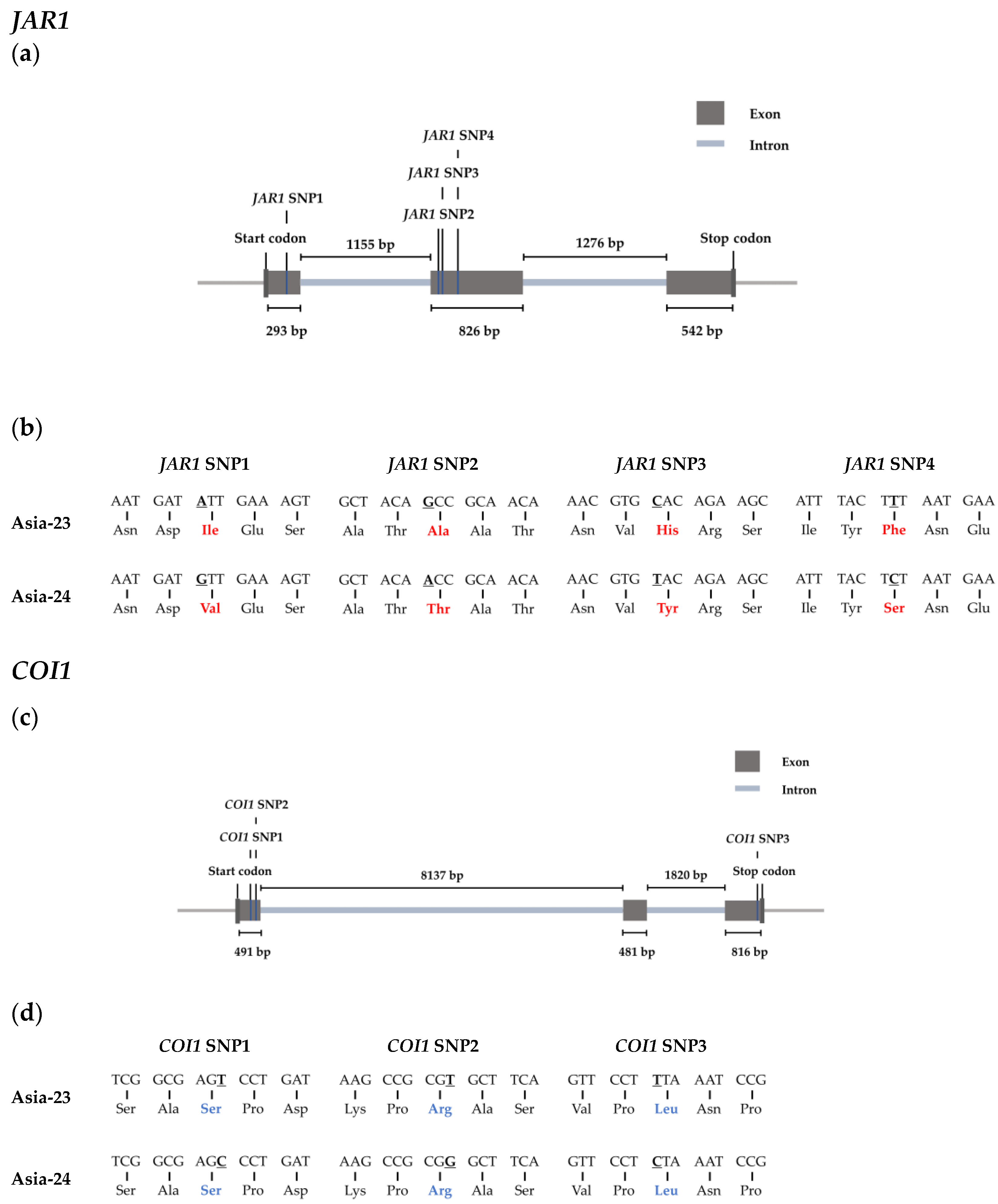
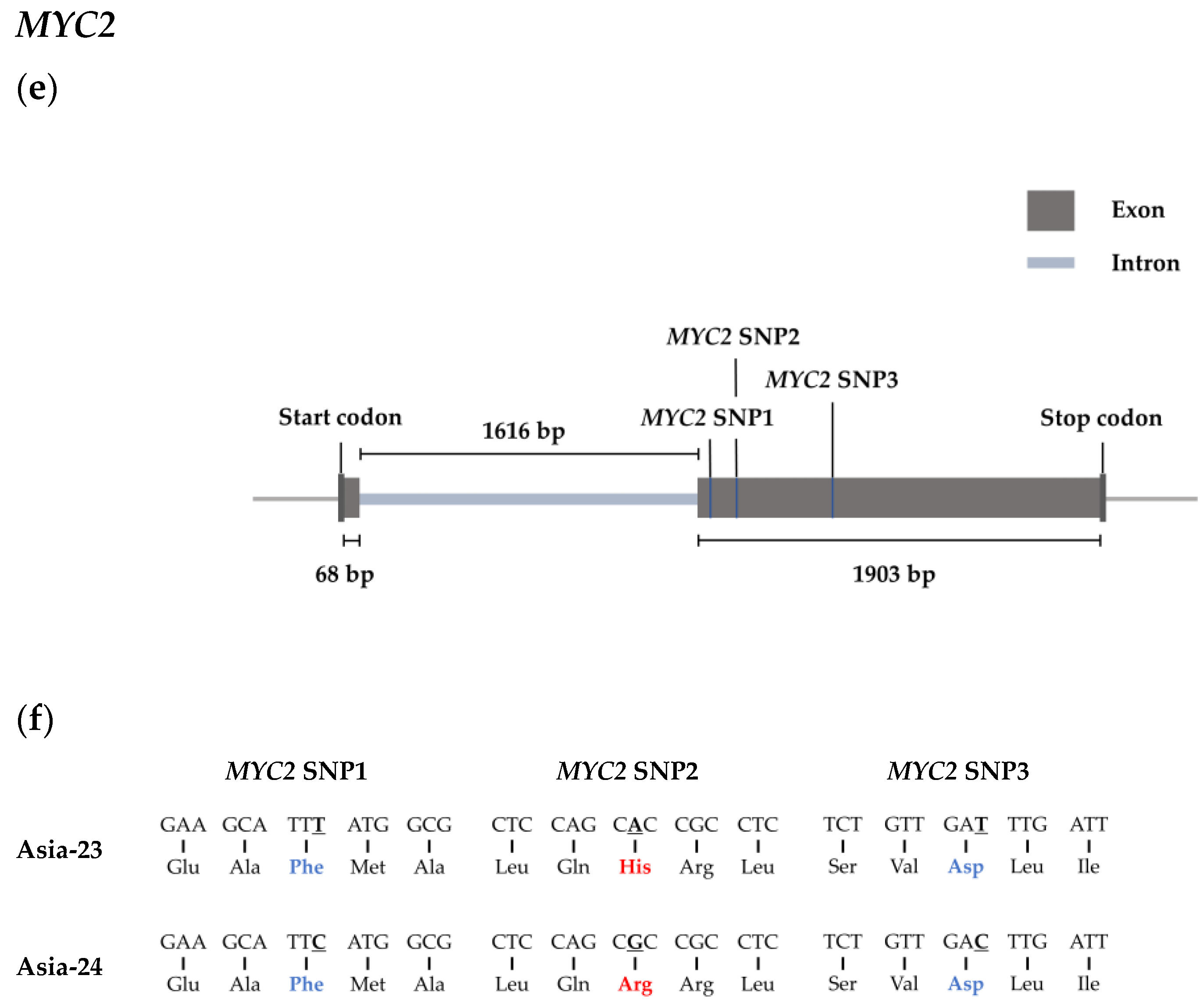
2.7. Selected Genes Sequence Confirmation from gDNA of Asia-23 and Asia-24
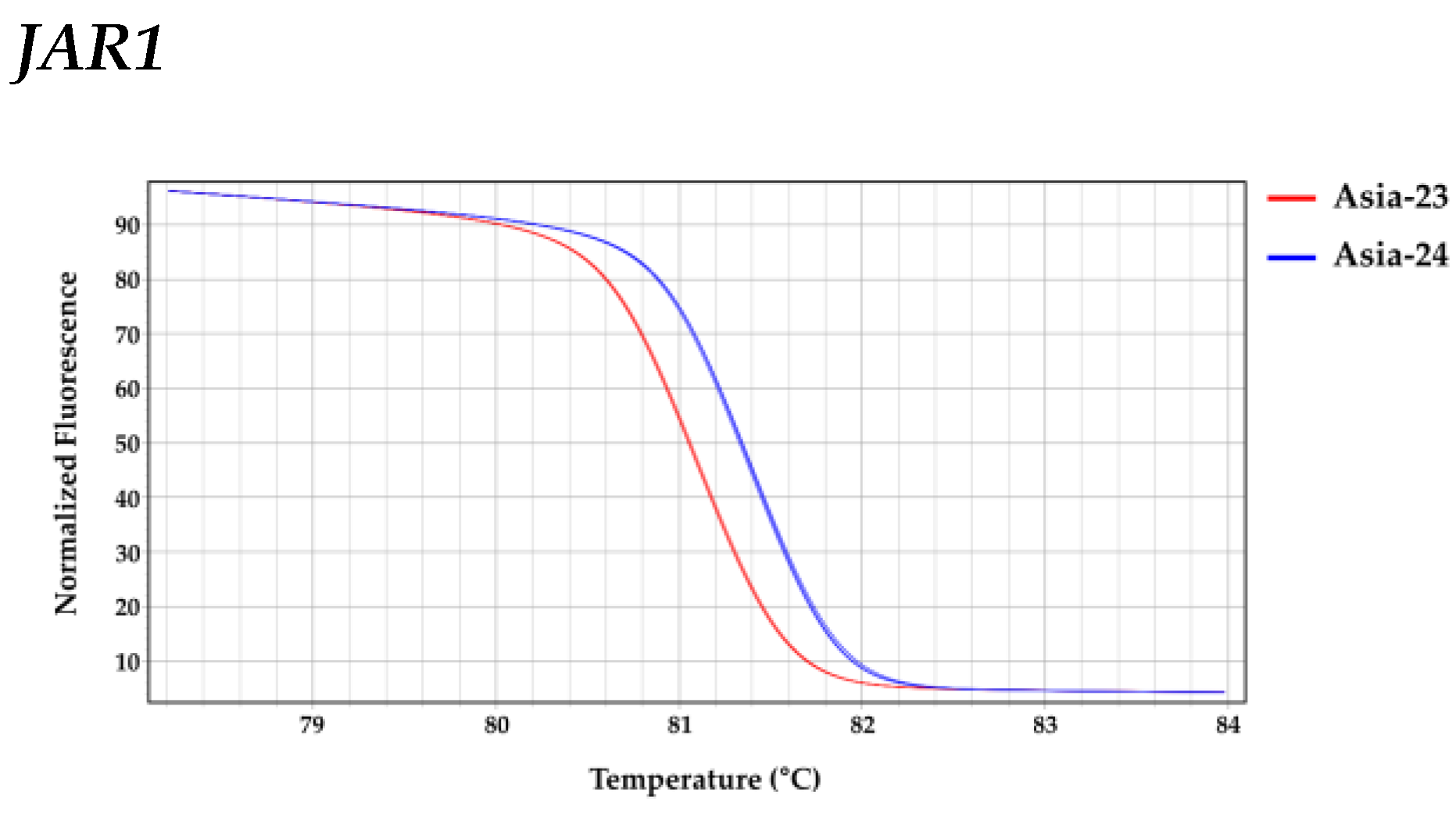
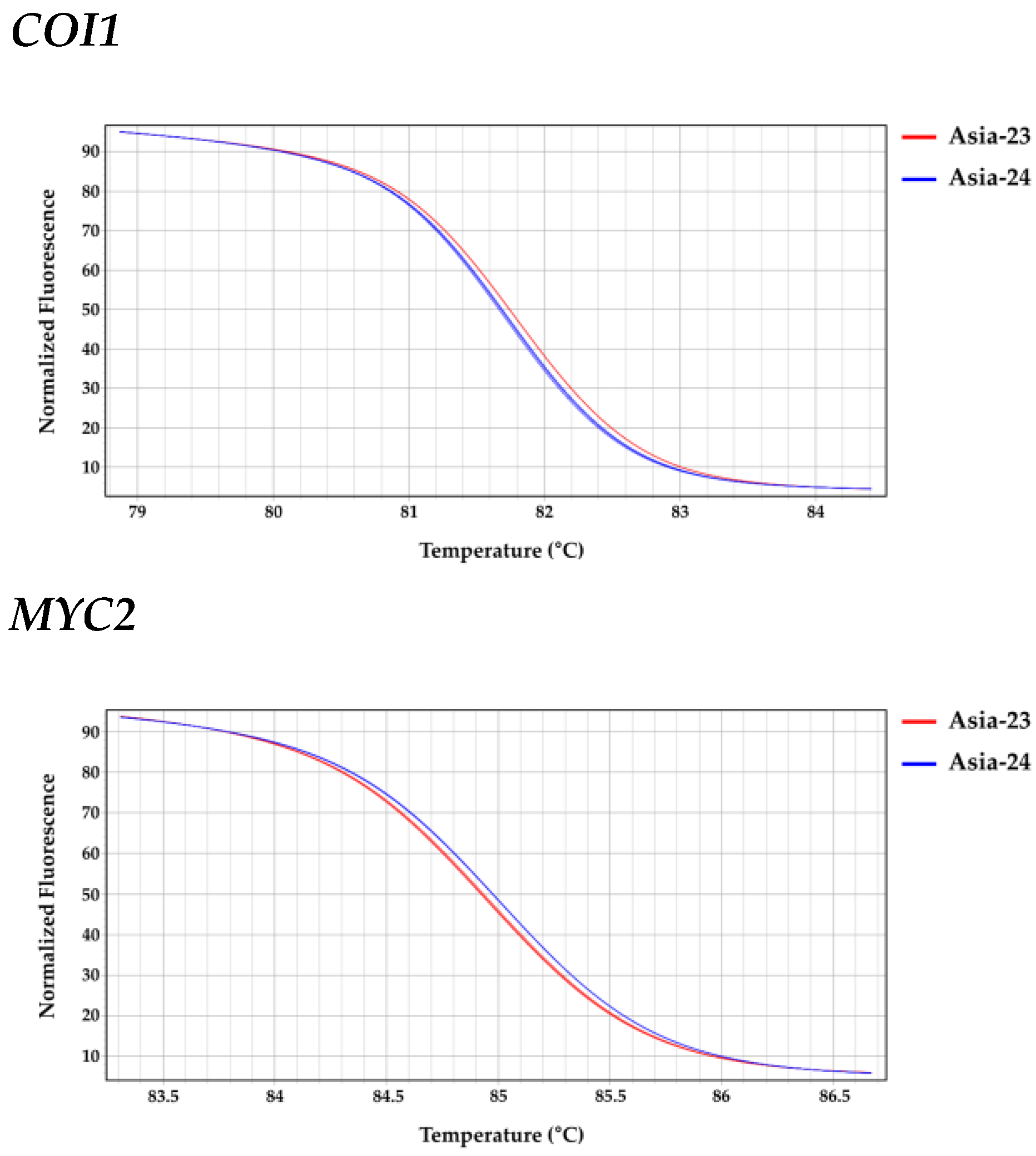
2.8. High Resolution Melting Analysis
3. Results
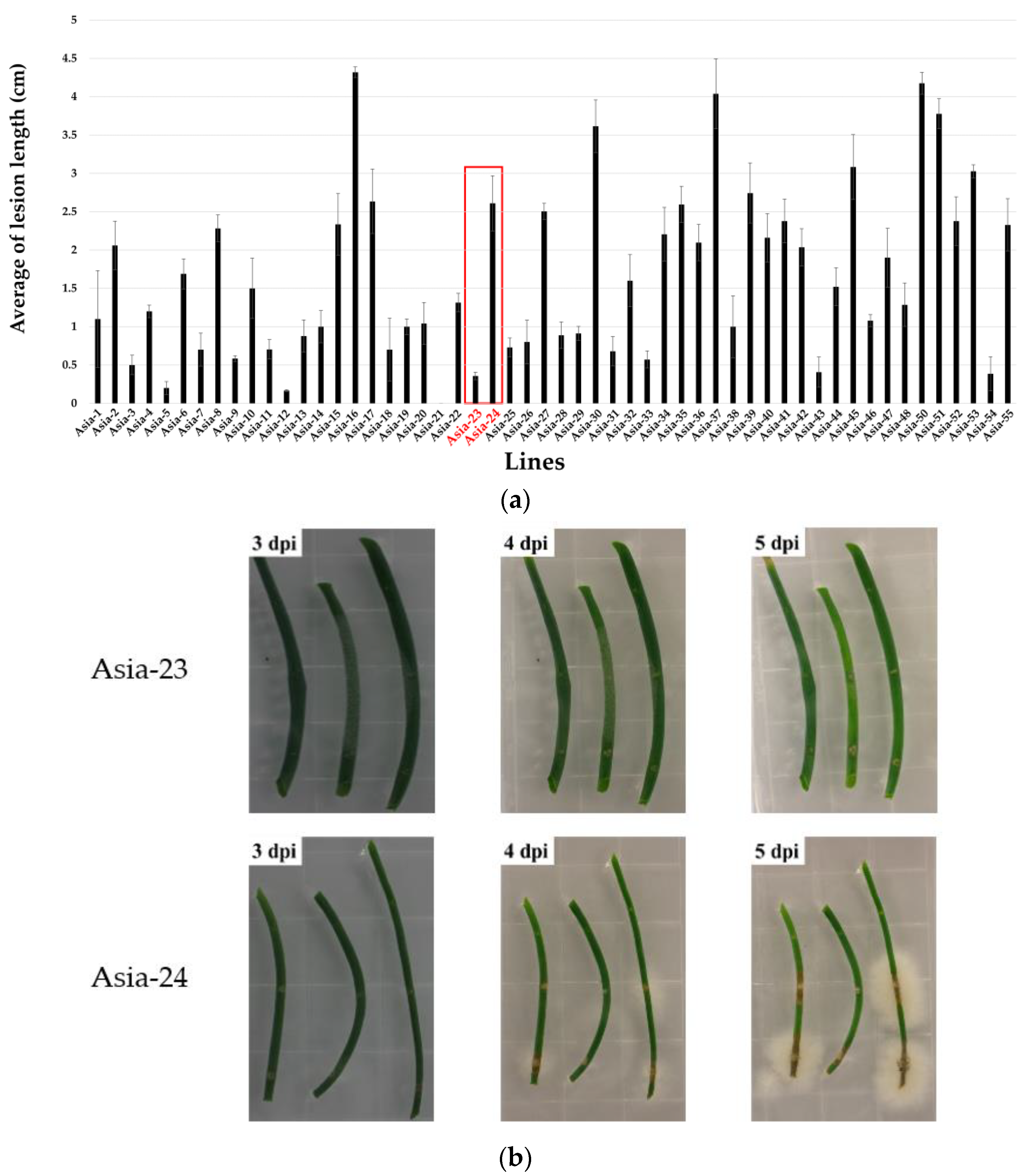
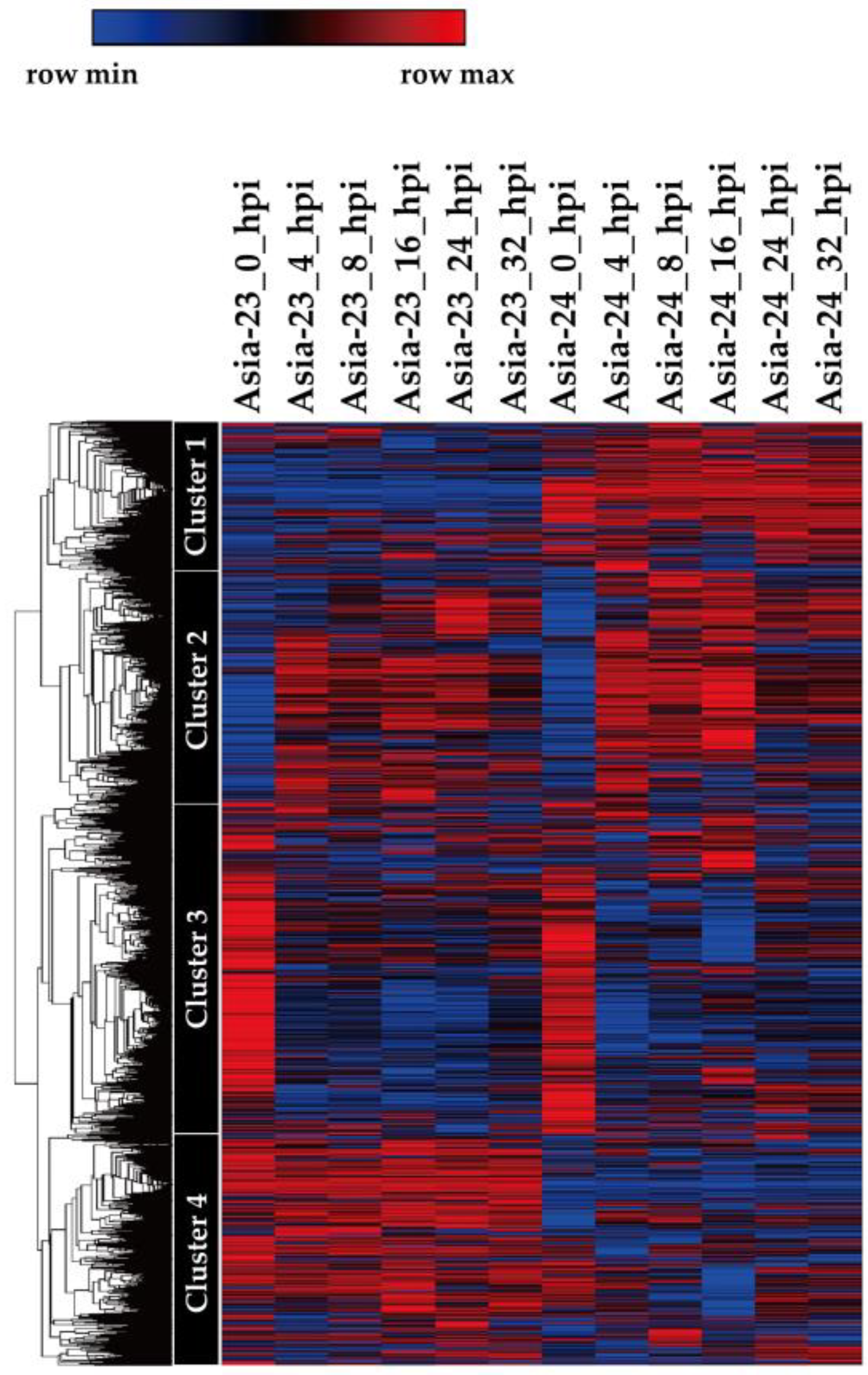
3.1. Screening Gray-Mold-Resistant and Susceptible Onions
3.2. RNA Sequencing and Sequence Data Pre-Processing
3.3. Differential Gene Expression Analysis
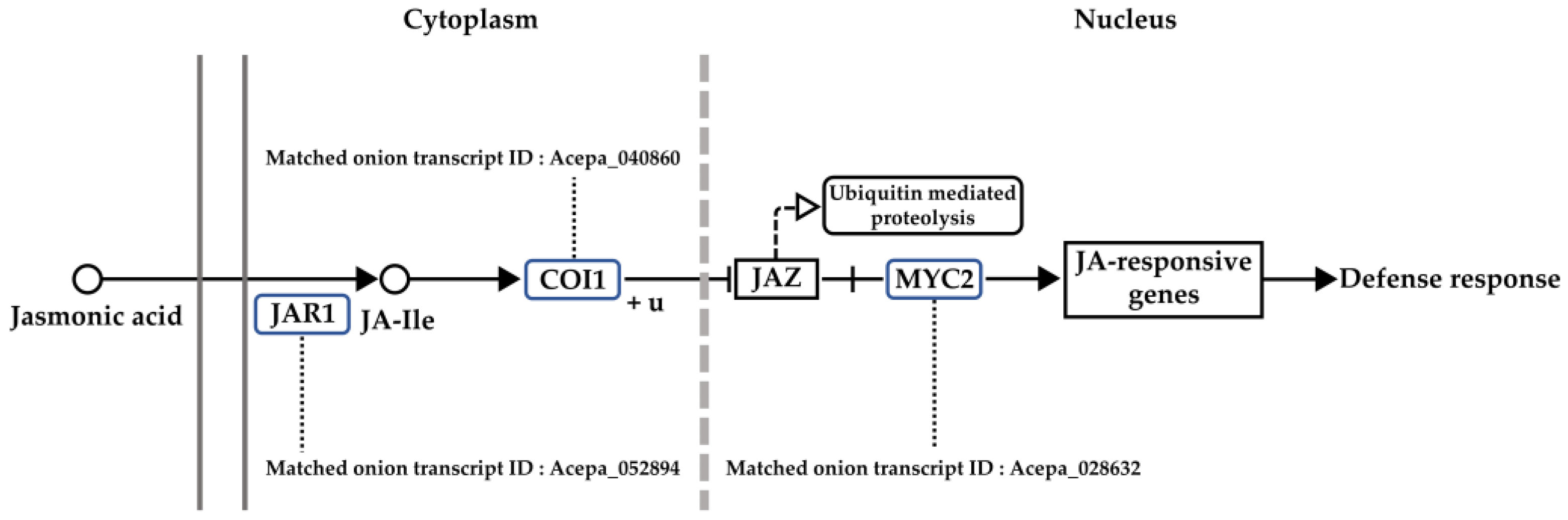
3.4. Analysis of Gene Network and Time-Series Expression
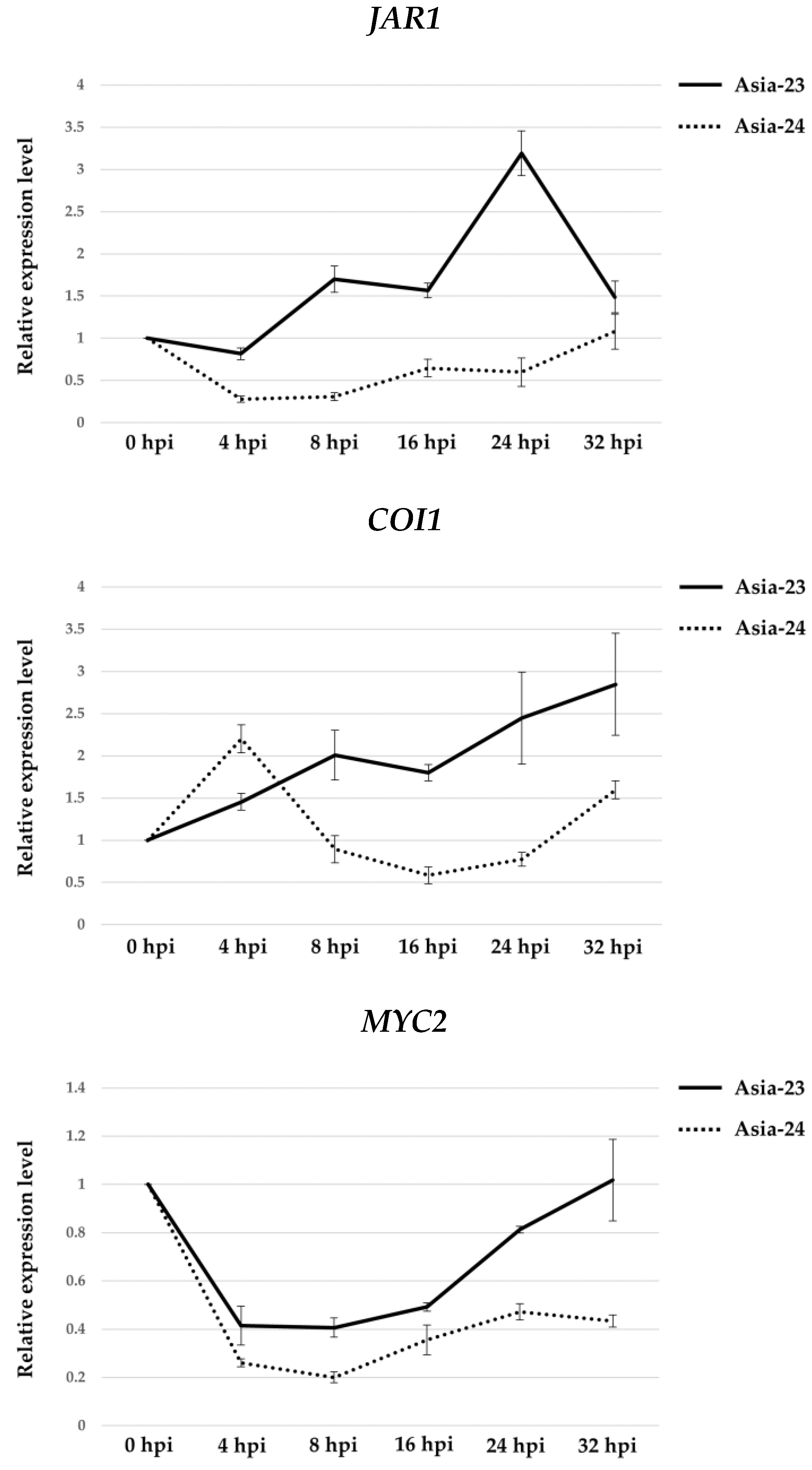
3.5. Verification of Gene Expression Using qRT-PCR
3.6. Confirmation of Selected Gene Sequences from gDNA of Asia-23 and Asia-24 and HRM Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van Rijswick, C. World Vegetable Map 2018: More Than Just a Local Affair. Available online: https://research.rabobank.com/far/en/sectors/regional-food-agri/world_vegetable_map_2018.html (accessed on 10 December 2021).
- FAOSTAT. Production/Yield Quantities of Onions, Dry in World + (Total). Available online: https://www.fao.org/faostat/en/#data/QCL/visualize (accessed on 10 December 2021).
- Yang, J.; Meyers, K.J.; van der Heide, J.; Liu, R.H. Varietal differences in phenolic content and antioxidant and antiproliferative activities of onions. J. Agric. Food. Chem. 2004, 52, 6787–6793. [Google Scholar] [CrossRef] [PubMed]
- Prakash, D.; Singh, B.N.; Upadhyay, G. Antioxidant and free radical scavenging activities of phenols from onion (Allium cepa). Food. Chem. 2007, 102, 1389–1393. [Google Scholar] [CrossRef]
- Albishi, T.; John, J.A.; Al-Khalifa, A.S.; Shahidi, F. Antioxidative Phenolic constituents of skins of onion varieties and their activities. J. Funct. Foods 2013, 5, 1191–1203. [Google Scholar] [CrossRef]
- Mlcek, J.; Valsikova, M.; Druzbikova, H.; Ryant, P.; Jurikova, T.; Sochor, J.; Borkovcova, M. The antioxidant capacity and macroelement content of several onion cultivars. Turk. J. Agric. For. 2015, 39, 999–1004. [Google Scholar] [CrossRef]
- Le Marchand, L. Cancer preventive effects of flavonoids—A review. Biomed. Pharmacother. 2002, 56, 296–301. [Google Scholar] [CrossRef]
- Galeone, C.; Pelucchi, C.; Levi, F.; Negri, E.; Franceschi, S.; Talamini, R.; Giacosa, A.; La Vecchia, C. Onion and garlic use and human cancer. Am. J. Clin. Nutr. 2006, 84, 1027–1032. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tian, W.X.; Ma, X.F. Inhibitory effects of onion (Allium cepa L.) extract on proliferation of cancer cells and adipocytes via inhibiting fatty acid synthase. Asian Pac. J. Cancer Prev. 2012, 13, 5573–5579. [Google Scholar] [CrossRef]
- Bisen, P.S.; Emerald, M. Nutritional and therapeutic potential of garlic and onion (Allium sp.). Curr. Nutr. Food Sci. 2016, 12, 190–199. [Google Scholar] [CrossRef]
- McCallum, J. Onion. In Vegetables; Kole, C., Ed.; Springer: Berlin/Heidelberg, Germany, 2007; Volume 5, pp. 331–347. [Google Scholar]
- Walker, J.C. Two undescribed species of Botrytis associated with the neck rot disease of onion bulbs. Phytopathology 1925, 15, 11. [Google Scholar]
- Hancock, J.G. Pathogenesis of Botrytis cinerea, B. squamosa, and B. allii on onion leaves. Phytopathology 1963, 53, 669–673. [Google Scholar]
- Lacy, M.L.; Lorbeer, J.W. Botrytis Neck Rot. In Compendium of Onion and Garlic Diseases; Schwartz, H.F., Mohan, S.K., Eds.; American Phytopathological Society: St Paul, MN, USA, 1995; pp. 18–19. [Google Scholar]
- Nielsen, K.; Yohalem, D.S.; Jensen, D.F. PCR detection and RFLP differentiation of Botrytis species associated with neck rot of onion. Plant Dis. 2002, 86, 682–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presly, A.H.; Maude, R.B. Control of Botrytis cinerea and Botrytis squamosa in overwintered salad onions by fungicide sprays. Ann. Appl. Biol. 1980, 94, 197–204. [Google Scholar] [CrossRef]
- Lorbeer, J.W.; Vincelli, P.C. Efficacy of dicarboximide fungicides and fungicide combinations for control of Botrytis leaf blight of onion in New York. Plant Dis. 1990, 74, 235–237. [Google Scholar] [CrossRef]
- Maude, R.B.; Shipway, M.R.; Presly, A.H.; O’connor, D. The effects of direct harvesting and drying systems on the incidence and control of neck rot (Botrytis allii) in onions. Plant Pathol. 1984, 33, 263–268. [Google Scholar] [CrossRef]
- Köhl, J.; Molhoek, W.W.L.; Goossen-Van De Geijn, H.M.; van der Plas, C.L. Potential of Ulocladium atrum for biocontrol of onion leaf spot through suppression of sporulation of Botrytis Spp. BioControl 2003, 48, 349–359. [Google Scholar] [CrossRef]
- Jorjandi, M.; Bonjar, G.S.; Baghizadeh, A.; Sirchi, G.S.; Massumi, H.; Baniasadi, F.; Aghighi, S.; Farokhi, P.R. Biocontrol of Botrytis allii Munn the causal agent of neck rot, the post harvest disease in onion, by use of a new Iranian isolate of Streptomyces. Am. J. Agric. Biol. Sci. 2009, 4, 72–78. [Google Scholar] [CrossRef] [Green Version]
- Currah, L.; Maude, R.B. Laboratory tests for leaf resistance to Botrytis squamosa in onions. Ann. Appl. Biol. 1984, 105, 277–283. [Google Scholar] [CrossRef]
- Lin, M.W.; Watson, J.F.; Baggett, J.R. Inheritance of resistance to neck-rot disease incited by Botrytis allii in bulb onions. J. Am. Soc. Hortic. Sci. 1995, 120, 297–299. [Google Scholar] [CrossRef] [Green Version]
- Walters, T.W.; Ellerbrock, L.A.; van der Heide, J.J.; Lorbeer, J.W.; LoParco, D.P. Field and greenhouse procedures to evaluate onions for Botrytis leaf blight resistance. HortScience 1996, 31, 436–438. [Google Scholar] [CrossRef] [Green Version]
- Anamika, K.; Verma, S.; Jere, A.; Desai, A. Transcriptomic profiling using next generation sequencing-advances, advantages, and challenges. In Next Generation Sequencing-Advances, Applications and Challenges; IntechOpen: Rijeka, Croatia, 2015; pp. 7355–7365. [Google Scholar]
- Khosa, J.S.; McCallum, J.; Dhatt, A.S.; Macknight, R.C. Enhancing onion breeding using molecular tools. Plant Breed. 2016, 135, 9–20. [Google Scholar] [CrossRef]
- Kim, S.; Kim, M.S.; Kim, Y.M.; Yeom, S.I.; Cheong, K.; Kim, K.T.; Jeon, J.; Kim, S.; Kim, D.S.; Sohn, S.H. Integrative structural annotation of de novo RNA-Seq provides an accurate reference gene set of the enormous genome of the onion (Allium cepa L.). DNA Res. 2015, 22, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Sohn, S.H.; Ahn, Y.K.; Lee, T.H.; Lee, J.E.; Jeong, M.H.; Seo, C.H.; Chandra, R.; Kwon, Y.S.; Kim, C.W.; Kim, D.S. Construction of a draft reference transcripts of onion (Allium cepa) using long-read sequencing. Plant Biotechnol. Rep. 2016, 10, 383–390. [Google Scholar] [CrossRef]
- Baek, G.; Kim, C.W.; Kim, S. Development of a molecular marker tightly linked to the C locus conferring a white bulb color in onion (Allium cepa L.) using bulked segregant analysis and RNA-Seq. Mol. Breed. 2017, 37, 94. [Google Scholar] [CrossRef]
- Jeon, S.; Han, J.; Kim, C.W.; Kim, J.G.; Moon, J.H.; Kim, S. Identification of a candidate gene responsible for the G locus determining chartreuse bulb color in onion (Allium cepa L.) using bulked segregant RNA-Seq. Theor. Appl. Genet. 2022, 1–22. [Google Scholar] [CrossRef]
- Zhang, C.; Li, X.; Zhan, Z.; Cao, L.; Zeng, A.; Chang, G.; Liang, Y. Transcriptome sequencing and metabolism analysis reveals the role of cyanidin metabolism in dark-red onion (Allium cepa L.) bulbs. Sci. Rep. 2018, 8, 14109. [Google Scholar] [CrossRef]
- Yuan, Q.; Song, C.; Gao, L.; Zhang, H.; Yang, C.; Sheng, J.; Ren, J.; Chen, D.; Wang, Y. Transcriptome de novo assembly and analysis of differentially expressed genes related to cytoplasmic male sterility in onion. Plant Physiol. Biochem. 2018, 125, 35–44. [Google Scholar] [CrossRef]
- Kim, S.; Kim, C.W.; Park, M.; Choi, D. Identification of candidate genes associated with fertility restoration of cytoplasmic male-sterility in onion (Allium cepa L.) using a combination of bulked segregant analysis and RNA-Seq. Theor. Appl. Genet. 2015, 128, 2289–2299. [Google Scholar] [CrossRef]
- Han, J.; Thamilarasan, S.K.; Natarajan, S.; Park, J.I.; Chung, M.Y.; Nou, I.S. De novo assembly and transcriptome analysis of bulb onion (Allium cepa L.) during cold acclimation using contrasting genotypes. PLoS ONE 2016, 11, e0161987. [Google Scholar]
- Ghodke, P.; Khandagale, K.; Thangasamy, A.; Kulkarni, A.; Narwade, N.; Shirsat, D.; Randive, P.; Roylawar, P.; Singh, I.; Gawande, S.J. Comparative transcriptome analyses in contrasting onion (Allium cepa L.) genotypes for drought stress. PLoS ONE 2020, 15, e0237457. [Google Scholar] [CrossRef]
- Maleck, K.; Dietrich, R.A. Defense on multiple fronts: How do plants cope with diverse enemies? Trends Plant Sci. 1999, 4, 215–219. [Google Scholar] [CrossRef]
- Howe, G.A.; Schilmiller, A.L. Oxylipin metabolism in response to stress. Curr. Opin. Plant Biol. 2002, 5, 230–236. [Google Scholar] [CrossRef]
- Gupta, A.; Hisano, H.; Hojo, Y.; Matsuura, T.; Ikeda, Y.; Mori, I.C.; Senthil-Kumar, M. Global profiling of phytohormone dynamics during combined drought and pathogen stress in Arabidopsis thaliana reveals ABA and JA as major regulators. Sci. Rep. 2017, 7, 4017. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Zhou, Y.; Zhou, M.; Yan, J.; Khurshid, M.; Weng, W.; Cheng, J.; Zhang, K. Jasmonic acid signaling pathway in plants. Int. J. Mol. Sci. 2019, 20, 2479. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Tian, S. Resistant responses of tomato fruit treated with exogenous methyl jasmonate to Botrytis cinerea infection. Sci. Hortic. 2012, 142, 38–43. [Google Scholar] [CrossRef]
- Wang, H.; Kou, X.; Wu, C.; Fan, G.; Li, T. Methyl jasmonate induces the resistance of postharvest blueberry to gray mold caused by Botrytis cinerea. J. Sci. Food Agric. 2020, 100, 4272–4281. [Google Scholar] [CrossRef] [PubMed]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Murashige, T.; Skoog, F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol. Plant 1962, 15, 473–497. [Google Scholar] [CrossRef]
- Abràmoff, M.D.; Magalhães, P.J.; Ram, S.J. Image processing with Image. J. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Park, J.S.; Park, T.; Lee, H.M.; Choi, J.R.; Park, Y.D. Development of molecular markers associated with resistance to gray mold disease in onion (Allium cepa L.) through RAPD-PCR and transcriptome analysis. Horticulturae 2021, 7, 436. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De novo transcript sequence reconstruction from RNA-Seq using the trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-Hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-Seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Nat. Preced. 2010, 11, R106. [Google Scholar]
- Swarbreck, D.; Wilks, C.; Lamesch, P.; Berardini, T.Z.; Garcia-Hernandez, M.; Foerster, H.; Li, D.; Meyer, T.; Muller, R.; Ploetz, L. The Arabidopsis Information Resource (TAIR): Gene structure and function annotation. Nucleic Acids Res. 2007, 36, D1009–D1014. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Finkers, R.; van Kaauwen, M.P.; Ament, K.; Burger-Meijer, K.; Egging, R.J.; Huits, H.; Kodde, L.P.; Kroon, L.; Shigyo, M.; Sato, S. Insights from the first genome assembly of onion (Allium cepa). bioRxiv 2021, 11, jkab243. [Google Scholar] [CrossRef]
- Rozen, S.; Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. In Bioinformatics Methods and Protocols; Misener, S., Krawetz, S.A., Eds.; Humana Press: Totowa, NJ, USA, 2000; Volume 132, pp. 365–386. [Google Scholar]
- Lee, G.H.; Lee, H.M.; Kim, S.J.; Park, Y.D. Development of DNA melting peak profile-based high-resolution melting (MP-HRM) analysis for genotyping germplasms in onion breeding. Hortic. Environ. Biotechnol. 2020, 61, 139–152. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzow, M.; Ragg, T. The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006, 7, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, O.; Lightfoot, S.; Schroeder, A. RNA integrity number (RIN)–standardization of RNA quality control. Agil. Appl. Note Publ. 2004, 1, 1–8. [Google Scholar]
- Liu, X.; Cao, X.; Shi, S.; Zhao, N.; Li, D.; Fang, P.; Chen, X.; Qi, W.; Zhang, Z. Comparative RNA-Seq analysis reveals a critical role for brassinosteroids in rose (Rosa hybrida) petal defense against Botrytis cinerea infection. BMC Genet. 2018, 19, 62. [Google Scholar] [CrossRef] [PubMed]
- Haile, Z.M.; Guzman, N.D.; Grace, E.; Moretto, M.; Sonego, P.; Engelen, K.; Zoli, L.; Moser, C.; Baraldi, E. Transcriptome profiles of strawberry (Fragaria vesca) fruit interacting with Botrytis cinerea at different ripening stages. Front. Plant Sci. 2019, 10, 1131. [Google Scholar] [CrossRef]
- Schouten, A.; Tenberge, K.B.; Vermeer, J.; Stewart, J.; Wagemakers, L.; Williamson, B.; Van Kan, J.A. Functional analysis of an extracellular catalase of Botrytis cinerea. Mol. Plant Pathol. 2002, 3, 227–238. [Google Scholar] [CrossRef]
- Wan, R.; Hou, X.; Wang, X.; Qu, J.; Singer, S.D.; Wang, Y.; Wang, X. Resistance evaluation of Chinese wild Vitis genotypes against Botrytis cinerea and different responses of resistant and susceptible hosts to the infection. Front. Plant Sci. 2015, 6, 854. [Google Scholar] [CrossRef]
- Asselbergh, B.; Curvers, K.; França, S.C.; Audenaert, K.; Vuylsteke, M.; Van Breusegem, F.; Höfte, M. Resistance to Botrytis cinerea in sitiens, an abscisic acid-deficient tomato mutant, involves timely production of hydrogen peroxide and cell wall modifications in the epidermis. Plant Physiol. 2007, 144, 1863–1877. [Google Scholar] [CrossRef] [Green Version]
- Staswick, P.E.; Tiryaki, I. The oxylipin signal jasmonic acid is activated by an enzyme that conjugates it to isoleucine in Arabidopsis. Plant Cell 2004, 16, 2117–2127. [Google Scholar] [CrossRef] [Green Version]
- Wakuta, S.; Suzuki, E.; Saburi, W.; Matsuura, H.; Nabeta, K.; Imai, R.; Matsui, H. OsJAR1 and OsJAR2 are jasmonyl-L-isoleucine synthases involved in wound-and pathogen-induced jasmonic acid signalling. Biochem. Biophys. Res. Commun. 2011, 409, 634–639. [Google Scholar] [CrossRef] [Green Version]
- Hui, S.; Hao, M.; Liu, H.; Xiao, J.; Li, X.; Yuan, M.; Wang, S. The group I GH3 family genes encoding JA-Ile synthetase act as positive regulator in the resistance of rice to Xanthomonas Oryzae pv. Oryzae. Biochem. Biophys. Res. Commun. 2019, 508, 1062–1066. [Google Scholar] [CrossRef]
- Xie, D.X.; Feys, B.F.; James, S.; Nieto-Rostro, M.; Turner, J.G. COI1: An Arabidopsis gene required for jasmonate-regulated defense and fertility. Science 1998, 280, 1091–1094. [Google Scholar] [CrossRef] [PubMed]
- Sheard, L.B.; Tan, X.; Mao, H.; Withers, J.; Ben-Nissan, G.; Hinds, T.R.; Kobayashi, Y.; Hsu, F.-F.; Sharon, M.; Browse, J. Jasmonate perception by inositol-phosphate-potentiated COI1–JAZ co-receptor. Nature 2010, 468, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Plotnikova, J.M.; De Lorenzo, G.; Ausubel, F.M. Arabidopsis local resistance to Botrytis cinerea involves salicylic acid and camalexin and requires EDS4 and PAD2, but not SID2, EDS5 or PAD4. Plant J. 2003, 35, 193–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomma, B.P.; Eggermont, K.; Penninckx, I.A.; Mauch-Mani, B.; Vogelsang, R.; Cammue, B.P.; Broekaert, W.F. Separate jasmonate-dependent and salicylate-dependent defense-response pathways in Arabidopsis are essential for resistance to distinct microbial pathogens. Proc. Natl. Acad. Sci. USA 1998, 95, 15107–15111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thatcher, L.F.; Manners, J.M.; Kazan, K. Fusarium oxysporum hijacks COI1-mediated jasmonate signaling to promote disease development in Arabidopsis. Plant J. 2009, 58, 927–939. [Google Scholar] [CrossRef]
- AbuQamar, S.; Chen, X.; Dhawan, R.; Bluhm, B.; Salmeron, J.; Lam, S.; Dietrich, R.A.; Mengiste, T. Expression profiling and mutant analysis reveals complex regulatory networks involved in Arabidopsis response to Botrytis infection. Plant J. 2006, 48, 28–44. [Google Scholar] [CrossRef]
- Lorenzo, O.; Chico, J.M.; Saénchez-Serrano, J.J.; Solano, R. JASMONATE-INSENSITIVE1 encodes a MYC transcription factor essential to discriminate between different jasmonate-regulated defense responses in Arabidopsis. Plant Cell 2004, 16, 1938–1950. [Google Scholar] [CrossRef] [Green Version]
- Du, M.; Zhao, J.; Tzeng, D.T.; Liu, Y.; Deng, L.; Yang, T.; Zhai, Q.; Wu, F.; Huang, Z.; Zhou, M. MYC2 orchestrates a hierarchical transcriptional cascade that regulates jasmonate-mediated plant immunity in tomato. Plant Cell 2017, 29, 1883–1906. [Google Scholar] [CrossRef] [Green Version]
- Weng, J.; Li, B.; Liu, C.; Yang, X.; Wang, H.; Hao, Z.; Li, M.; Zhang, D.; Ci, X.; Li, X. A non-synonymous SNP within the isopentenyl transferase 2 locus is associated with kernel weight in Chinese maize inbreds (Zea Mays L.). BMC Plant Biol. 2013, 13, 98. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.J.; Seo, M.; Hersh, C.P.; Rhee, S.J.; Kim, Y.; Lee, G.P. An evolutionarily conserved non-synonymous SNP in a leucine-rich repeat domain determines anthracnose resistance in watermelon. Theor. Appl. Genet. 2019, 132, 473–488. [Google Scholar] [CrossRef]
- Shabalina, S.A.; Spiridonov, N.A.; Kashina, A. Sounds of silence: Synonymous nucleotides as a key to biological regulation and complexity. Nucleic Acids Res. 2013, 41, 2073–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bark, O.H.; Havey, M.J. Similarities and relationships among populations of the bulb onion as estimated by nuclear RFLPs. Theor. Appl. Genet. 1995, 90, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Tanikawa, T.; Takagi, M.; Ichii, M. Cultivar identification and genetic diversity in onion (Allium cepa L.) as evaluated by random amplified polymorphic DNA (RAPD) analysis. J. Jpn. Soc. Hortic. Sci. 2002, 71, 249–251. [Google Scholar] [CrossRef]
- van Heusden, A.W.; Shigyo, M.; Tashiro, Y.; Vrielink-van Ginkel, R.; Kik, C. AFLP linkage group assignment to the chromosomes of Allium cepa L. via monosomic addition lines. Theor. Appl. Genet. 2000, 100, 480–486. [Google Scholar] [CrossRef]
- McCallum, J.; Thomson, S.; Pither-Joyce, M.; Kenel, F.; Clarke, A.; Havey, M.J. Genetic diversity analysis and single-nucleotide polymorphism marker development in cultivated bulb onion based on expressed sequence tag–simple sequence repeat markers. J. Am. Soc. Hortic. Sci. 2008, 133, 810–818. [Google Scholar] [CrossRef] [Green Version]
- Duangjit, J.; Bohanec, B.; Chan, A.P.; Town, C.D.; Havey, M.J. Transcriptome sequencing to produce SNP-based genetic maps of onion. Theor. Appl. Genet. 2013, 126, 2093–2101. [Google Scholar] [CrossRef]
- Scholten, O.E.; van Kaauwen, M.P.; Shahin, A.; Hendrickx, P.M.; Keizer, L.P.; Burger, K.; van Heusden, A.W.; van der Linden, C.G.; Vosman, B. SNP-markers in Allium species to facilitate introgression breeding in onion. BMC Plant Biol. 2016, 16, 187. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Line | LS Means (Melting Peak Tm (°C)) | Groups * | |
|---|---|---|---|---|
| JAR1 | Asia-24 | 81.387 | A | |
| Asia-23 | 81.093 | B | ||
| COI1 | Asia-23 | 81.783 | A | |
| Asia-24 | 81.730 | B | ||
| MYC2 | Asia-24 | 85.007 | A | |
| Asia-23 | 84.953 | B | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-M.; Park, J.-S.; Kim, S.-J.; Kim, S.-G.; Park, Y.-D. Using Transcriptome Analysis to Explore Gray Mold Resistance-Related Genes in Onion (Alliumcepa L.). Genes 2022, 13, 542. https://doi.org/10.3390/genes13030542
Lee H-M, Park J-S, Kim S-J, Kim S-G, Park Y-D. Using Transcriptome Analysis to Explore Gray Mold Resistance-Related Genes in Onion (Alliumcepa L.). Genes. 2022; 13(3):542. https://doi.org/10.3390/genes13030542
Chicago/Turabian StyleLee, Hyun-Min, Jee-Soo Park, So-Jeong Kim, Seung-Gyu Kim, and Young-Doo Park. 2022. "Using Transcriptome Analysis to Explore Gray Mold Resistance-Related Genes in Onion (Alliumcepa L.)" Genes 13, no. 3: 542. https://doi.org/10.3390/genes13030542