
Comparative Analysis of microRNA Binding Site Distribution and microRNA-Mediated Gene Expression Repression of Oncogenes and Tumor Suppressor Genes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Evolutionarily Conserved miRNA Binding Sites
2.2. Comparative RNA-seq Analysis of Wildtype and Dicer1 Knockout (KO) Cells
2.3. Comparative Polysome Profiling Analysis of Wildtype and Dicer1 KO HCT116 Cells
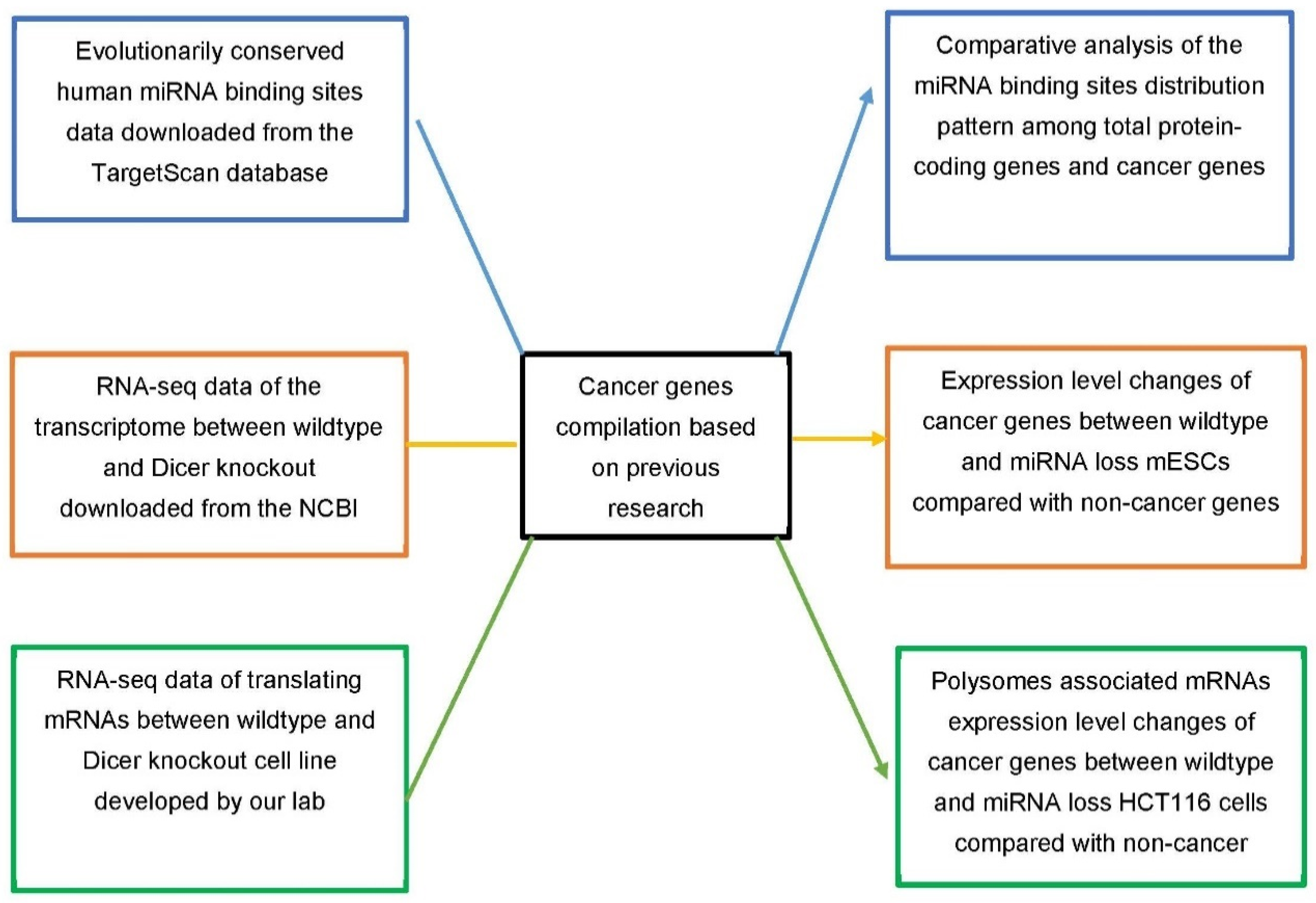
2.4. Compilation of Cancer Genes and miRNA Binding Sites, and Analysis of mRNA Polysome Association (Translation Activity) in miRNA-Production-Deficient Cells
2.5. Computer Software
2.6. Overall Study Design
3. Results
3.1. Distribution of miRNA Binding Sites among Cancer Genes
3.2. The TSG mRNA Abundances Are Depressed More than Oncogene mRNA Abundances in DICER1 KO mESCs
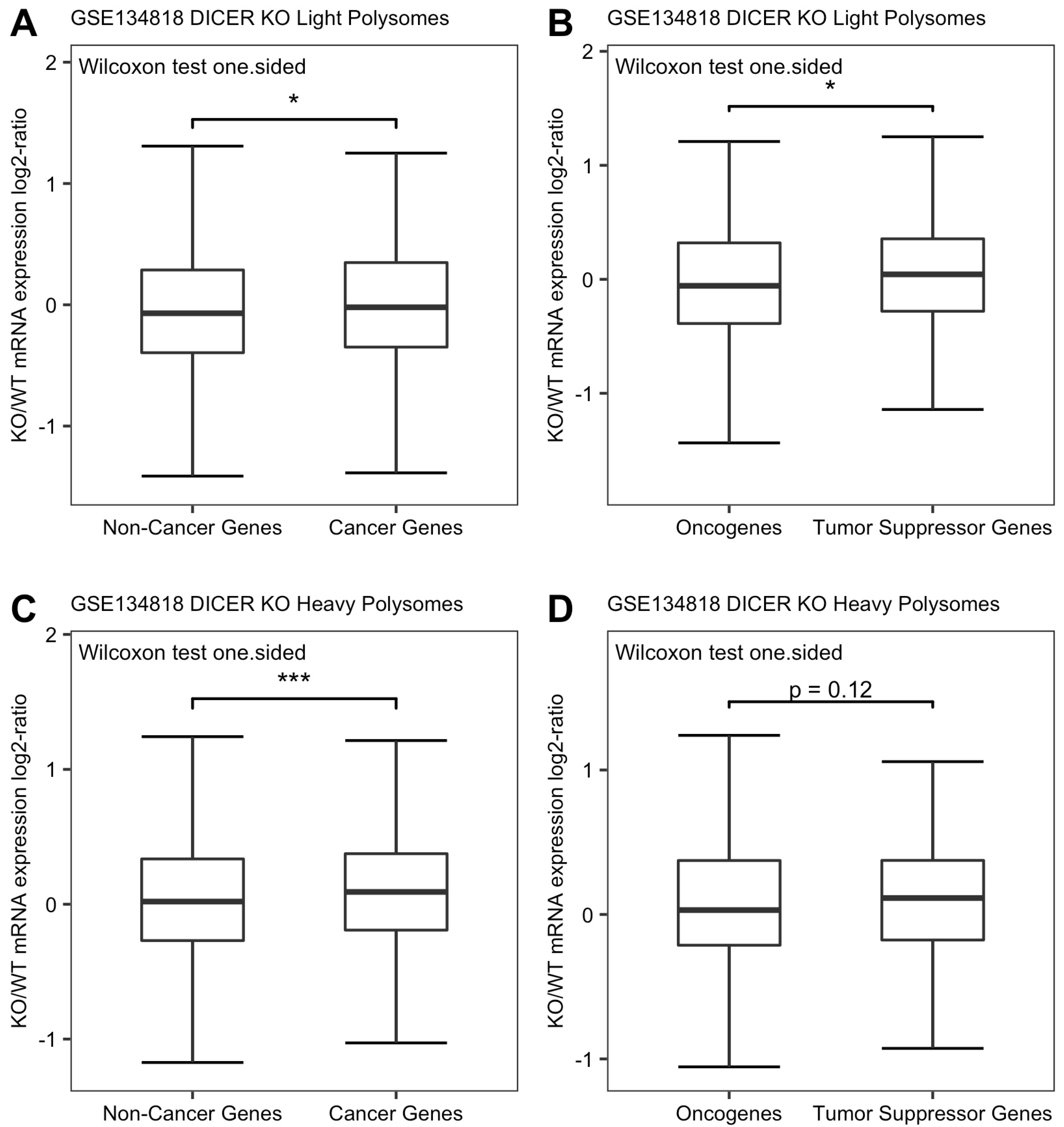
3.3. Translating TSG mRNA Abundances Are Depressed More than Those of Oncogene mRNAs in Dicer Knockout Human HCT116 Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Molnár, A.; Schwach, F.; Studholme, D.J.; Thuenemann, E.C.; Baulcombe, D.C. miRNAs control gene expression in the single-cell alga Chlamydomonas reinhardtii. Nature 2007, 447, 1126–1129. [Google Scholar] [CrossRef] [PubMed]
- Melo, C.A.; Melo, S.A. Biogenesis and Physiology of MicroRNAs. In Non-Coding RNAs and Cancer; Fabbri, M., Ed.; Springer: New York, NY, USA, 2014; pp. 5–24. [Google Scholar]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [Green Version]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Selbach, M.; Schwanhausser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread changes in protein synthesis induced by microRNAs. Nature 2008, 455, 58–63. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Quillet, A.; Saad, C.; Ferry, G.; Anouar, Y.; Vergne, N.; Lecroq, T.; Dubessy, C. Improving bioinformatics prediction of microRNA targets by ranks aggregation. Front. Genet. 2020, 10, 1330. [Google Scholar] [CrossRef] [Green Version]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebert, L.F.R.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef] [PubMed]
- DeVeale, B.; Swindlehurst-Chan, J.; Blelloch, R. The roles of microRNAs in mouse development. Nat. Rev. Genet. 2021, 22, 307–323. [Google Scholar] [CrossRef] [PubMed]
- Nigi, L.; Grieco, G.E.; Ventriglia, G.; Brusco, N.; Mancarella, F.; Formichi, C.; Dotta, F.; Sebastiani, G. MicroRNAs as regulators of insulin signaling: Research updates and potential therapeutic perspectives in type 2 diabetes. Int. J. Mol. Sci. 2018, 19, 3705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Im, H.-I.; Kenny, P.J. MicroRNAs in neuronal function and dysfunction. Trends Neurosci. 2012, 35, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Rottiers, V.; Näär, A.M. MicroRNAs in metabolism and metabolic disorders. Nat. Rev. Mol. Cell Biol. 2012, 13, 239–250. [Google Scholar] [CrossRef]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Hayes, J.; Peruzzi, P.P.; Lawler, S. MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol. Med. 2014, 20, 460–469. [Google Scholar] [CrossRef]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Qiu, C.; Zhang, H.; Wang, J.; Cui, Q.; Yin, Y. Human microRNA oncogenes and tumor suppressors show significantly different biological patterns: From functions to targets. PLoS ONE 2010, 5, e13067. [Google Scholar] [CrossRef]
- Zhang, B.; Pan, X.; Cobb, G.P.; Anderson, T.A. microRNAs as oncogenes and tumor suppressors. Dev. Biol. 2007, 302, 1768–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, A.; Jacks, T. MicroRNAs and cancer: Short RNAs go a long way. Cell 2009, 136, 586–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Leva, G.; Garofalo, M.; Croce, C.M. MicroRNAs in cancer. Annu. Rev. Pathol. 2014, 9, 287–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svoronos, A.A.; Engelman, D.M.; Slack, F.J. OncomiR or Tumor Suppressor? The Duplicity of MicroRNAs in Cancer. Cancer Res. 2016, 76, 3666–3670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Oh, S.; Abdelnabi, J.; Al-Dulaimi, R.; Aggarwal, A.; Ramos, M.; Davis, S.; Riester, M.; Waldron, L. HGNChelper: Identification and correction of invalid gene symbols for human and mouse [version 1; peer review: 2 approved, 1 approved with reservations]. F1000Research 2020, 9, 1490. [Google Scholar] [CrossRef]
- Zheng, G.X.; Do, B.T.; Webster, D.E.; Khavari, P.A.; Chang, H.Y. Dicer-microRNA-Myc circuit promotes transcription of hundreds of long noncoding RNAs. Nat. Struct. Mol. Biol. 2014, 21, 585–590. [Google Scholar] [CrossRef] [Green Version]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wang, T.; Gill, A.; Hilliard, T.; Chen, F.; Karamyshev, A.L.; Zhang, F. Uncovering the cellular capacity for intensive and specific feedback self-control of the argonautes and MicroRNA targeting activity. Nucleic Acids Res. 2020, 48, 4681–4697. [Google Scholar] [CrossRef] [Green Version]
- Sack, L.M.; Davoli, T.; Li, M.Z.; Li, Y.; Xu, Q.; Naxerova, K.; Wooten, E.C.; Bernardi, R.J.; Martin, T.D.; Chen, T. Profound tissue specificity in proliferation control underlies cancer drivers and aneuploidy patterns. Cell 2018, 173, 499–514.e423. [Google Scholar] [CrossRef] [Green Version]
- Davoli, T.; Xu, A.W.; Mengwasser, K.E.; Sack, L.M.; Yoon, J.C.; Park, P.J.; Elledge, S.J. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 2013, 155, 948–962. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Barabasi, A.L.; Albert, R. Emergence of scaling in random networks. Science 1999, 286, 509–512. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Jiang, W.; Lages, N.; Borcherds, W.; Wang, D. Relationship between gene duplicability and diversifiability in the topology of biochemical networks. BMC Genom. 2014, 15, 577. [Google Scholar] [CrossRef] [Green Version]
- Padawer, T.; Leighty, R.E.; Wang, D. Duplicate gene enrichment and expression pattern diversification in multicellularity. Nucleic Acids Res. 2012, 40, 7597–7605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Wang, D. The Pattern of microRNA Binding Site Distribution. Genes 2017, 8, 296. [Google Scholar] [CrossRef] [Green Version]
- Zare, H.; Khodursky, A.; Sartorelli, V. An evolutionarily biased distribution of miRNA sites toward regulatory genes with high promoter-driven intrinsic transcriptional noise. BMC Evol. Biol. 2014, 14, 74. [Google Scholar] [CrossRef] [Green Version]
- Shalgi, R.; Lieber, D.; Oren, M.; Pilpel, Y. Global and local architecture of the mammalian microRNA–transcription factor regulatory network. PLoS Comput. Biol. 2007, 3, e131. [Google Scholar] [CrossRef]
- Hon, L.S.; Zhang, Z. The roles of binding site arrangement and combinatorial targeting in microRNA repression of gene expression. Genome Biol 2007, 8, R166. [Google Scholar] [CrossRef] [Green Version]
- Pasquinelli, A.E. MicroRNAs and their targets: Recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 2012, 13, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Chassé, H.; Boulben, S.; Costache, V.; Cormier, P.; Morales, J. Analysis of translation using polysome profiling. Nucleic Acids Res. 2017, 45, e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandin, V.; Sikström, K.; Alain, T.; Morita, M.; McLaughlan, S.; Larsson, O.; Topisirovic, I. Polysome fractionation and analysis of mammalian translatomes on a genome-wide scale. J. Vis. Exp. JoVE 2014, 17, 51455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, H.A.; Gerber, A.P. Translatome profiling: Methods for genome-scale analysis of mRNA translation. Brief. Funct. Genom. 2016, 15, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Bracken, C.P.; Scott, H.S.; Goodall, G.J. A network-biology perspective of microRNA function and dysfunction in cancer. Nat. Rev. Genet 2016, 17, 719–732. [Google Scholar] [CrossRef]
- Papagiannakopoulos, T.; Shapiro, A.; Kosik, K.S. MicroRNA-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Res. 2008, 68, 8164–8172. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Liao, Y.; Tang, L. MicroRNA-34 family: A potential tumor suppressor and therapeutic candidate in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 53. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Wang, Z.; Fillmore, R.; Xi, Y. MiR-200, a new star miRNA in human cancer. Cancer Lett. 2014, 344, 166–173. [Google Scholar] [CrossRef] [Green Version]
- Jadideslam, G.; Ansarin, K.; Sakhinia, E.; Babaloo, Z.; Abhari, A.; Ghahremanzadeh, K.; Khalili, M.; Radmehr, R.; Kabbazi, A. Diagnostic biomarker and therapeutic target applications of miR-326 in cancers: A systematic review. J. Cell. Physiol. 2019, 234, 21560–21574. [Google Scholar] [CrossRef]
- Serra, F.; Bottini, S.; Pratella, D.; Stathopoulou, M.G.; Sebille, W.; El-Hami, L.; Repetto, E.; Mauduit, C.; Benahmed, M.; Grandjean, V.; et al. Systemic CLIP-seq analysis and game theory approach to model microRNA mode of binding. Nucleic Acids Res. 2021, 49, e66. [Google Scholar] [CrossRef]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichhorn, S.W.; Guo, H.; McGeary, S.E.; Rodriguez-Mias, R.A.; Shin, C.; Baek, D.; Hsu, S.-H.; Ghoshal, K.; Villén, J.; Bartel, D.P. mRNA destabilization is the dominant effect of mammalian microRNAs by the time substantial repression ensues. Mol. Cell 2014, 56, 104–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazzini, A.A.; Lee, M.T.; Giraldez, A.J. Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science 2012, 336, 233–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Béthune, J.; Artus-Revel, C.G.; Filipowicz, W. Kinetic analysis reveals successive steps leading to miRNA-mediated silencing in mammalian cells. EMBO Rep. 2012, 13, 716–723. [Google Scholar] [CrossRef] [Green Version]
- Djuranovic, S.; Nahvi, A.; Green, R. miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science 2012, 336, 237–240. [Google Scholar] [CrossRef] [Green Version]
- Freimer, J.W.; Hu, T.J.; Blelloch, R. Decoupling the impact of microRNAs on translational repression versus RNA degradation in embryonic stem cells. Elife 2018, 7, e38014. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, S.; Wang, J.; Zhang, F.; Wang, D. Comparative Analysis of microRNA Binding Site Distribution and microRNA-Mediated Gene Expression Repression of Oncogenes and Tumor Suppressor Genes. Genes 2022, 13, 481. https://doi.org/10.3390/genes13030481
Tian S, Wang J, Zhang F, Wang D. Comparative Analysis of microRNA Binding Site Distribution and microRNA-Mediated Gene Expression Repression of Oncogenes and Tumor Suppressor Genes. Genes. 2022; 13(3):481. https://doi.org/10.3390/genes13030481
Chicago/Turabian StyleTian, Shuangmei, Jing Wang, Fangyuan Zhang, and Degeng Wang. 2022. "Comparative Analysis of microRNA Binding Site Distribution and microRNA-Mediated Gene Expression Repression of Oncogenes and Tumor Suppressor Genes" Genes 13, no. 3: 481. https://doi.org/10.3390/genes13030481