
Epigenetic and Epitranscriptomic Control in Prostate Cancer

 , and
, and
Abstract
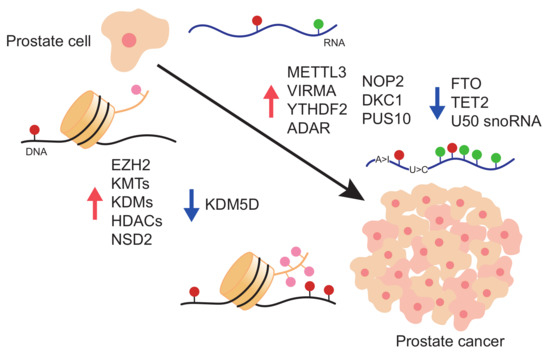
:
1. Prostate Cancer
2. Epigenetic Alterations in Prostate Cancer
2.1. Writers
2.2. Readers
2.3. Erasers
3. Epitranscriptomics Alterations in Prostate Cancer
3.1. m6A Deposition in RNA and Its Role in PCa
3.2. m5C and hm5C Deposition in RNA and Its Role in PCa
3.3. Pseudouridine in RNA and Its Role in PCa
3.4. RNA Editing in PCa
3.5. 2′-O-methylation in PCa
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernar, C.H.; Ebot, E.M.; Wilson, K.M.; Mucci, L.A. The Epidemiology of Prostate Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a030361. [Google Scholar] [CrossRef] [PubMed]
- Schatten, H. Brief Overview of Prostate Cancer Statistics, Grading, Diagnosis and Treatment Strategies. Adv. Exp. Med. Biol. 2018, 1095, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nunzio, C.D.E.; Presicce, F.; Giacinti, S.; Bassanelli, M.; Tubaro, A. Castration-resistance prostate cancer: What is in the pipeline? Minerva Urol. Nefrol. 2018, 70, 22–41. [Google Scholar] [CrossRef]
- Armstrong, C.M.; Gao, A.C. Adaptive pathways and emerging strategies overcoming treatment resistance in castration resistant prostate cancer. Asian J. Urol. 2016, 3, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The genomic complexity of primary human prostate cancer. Nature 2011, 470, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [Green Version]
- Abate-Shen, C.; Shen, M.M. Molecular genetics of prostate cancer. Genes Dev. 2000, 14, 2410–2434. [Google Scholar] [CrossRef] [Green Version]
- Abida, W.; Armenia, J.; Gopalan, A.; Brennan, R.; Walsh, M.; Barron, D.; Danila, D.; Rathkopf, D.; Morris, M.; Slovin, S.; et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis. Oncol. 2017, 2017, PO.17.00029. [Google Scholar] [CrossRef]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, T.W.; Roy, R.; Tomlins, S.A.; Ngo, V.T.; Kobayashi, Y.; Azameera, A.; Rubin, M.A.; Pienta, K.J.; Chinnaiyan, A.; Ittmann, M.M.; et al. Common structural and epigenetic changes in the genome of castration-resistant prostate cancer. Cancer Res. 2012, 72, 616–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonpavde, G.; Aparicio, A.M.; Zhan, F.; North, B.; Delaune, R.; Garbo, L.E.; Rousey, S.R.; Weinstein, R.E.; Xiao, L.; Boehm, K.A.; et al. Azacitidine favorably modulates PSA kinetics correlating with plasma DNA LINE-1 hypomethylation in men with chemonaïve castration-resistant prostate cancer. Urol. Oncol. 2011, 29, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Singal, R.; Ramachandran, K.; Gordian, E.; Quintero, C.; Zhao, W.; Reis, I.M. Phase I/II study of azacitidine, docetaxel, and prednisone in patients with metastatic castration-resistant prostate cancer previously treated with docetaxel-based therapy. Clin. Genitourin. Cancer 2015, 13, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.R.; Schweizer, M.T.; Nanus, D.M.; Pantuck, A.J.; Heath, E.I.; Campeau, E.; Attwell, S.; Norek, K.; Snyder, M.; Bauman, L.; et al. A Phase Ib/IIa Study of the Pan-BET Inhibitor ZEN-3694 in Combination with Enzalutamide in Patients with Metastatic Castration-resistant Prostate Cancer. Clin. Cancer Res. 2020, 26, 5338–5347. [Google Scholar] [CrossRef]
- Piha-Paul, S.A.; Sachdev, J.C.; Barve, M.; LoRusso, P.; Szmulewitz, R.; Patel, S.P.; Lara, P.N., Jr.; Chen, X.; Hu, B.; Freise, K.J.; et al. First-in-Human Study of Mivebresib (ABBV-075), an Oral Pan-Inhibitor of Bromodomain and Extra Terminal Proteins, in Patients with Relapsed/Refractory Solid Tumors. Clin. Cancer Res. 2019, 25, 6309–6319. [Google Scholar] [CrossRef] [Green Version]
- Lewin, J.; Soria, J.C.; Stathis, A.; Delord, J.P.; Peters, S.; Awada, A.; Aftimos, P.G.; Bekradda, M.; Rezai, K.; Zeng, Z.; et al. Phase Ib Trial With Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients With Selected Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 3007–3014. [Google Scholar] [CrossRef]
- Bradley, D.; Rathkopf, D.; Dunn, R.; Stadler, W.M.; Liu, G.; Smith, D.C.; Pili, R.; Zwiebel, J.; Scher, H.; Hussain, M. Vorinostat in advanced prostate cancer patients progressing on prior chemotherapy (National Cancer Institute Trial 6862): Trial results and interleukin-6 analysis: A study by the Department of Defense Prostate Cancer Clinical Trial Consortium and University of Chicago Phase 2 Consortium. Cancer 2009, 115, 5541–5549. [Google Scholar] [CrossRef] [Green Version]
- Schneider, B.J.; Kalemkerian, G.P.; Bradley, D.; Smith, D.C.; Egorin, M.J.; Daignault, S.; Dunn, R.; Hussain, M. Phase I study of vorinostat (suberoylanilide hydroxamic acid, NSC 701852) in combination with docetaxel in patients with advanced and relapsed solid malignancies. Investig. New Drugs 2012, 30, 249–257. [Google Scholar] [CrossRef]
- Wheler, J.J.; Janku, F.; Falchook, G.S.; Jackson, T.L.; Fu, S.; Naing, A.; Tsimberidou, A.M.; Moulder, S.L.; Hong, D.S.; Yang, H.; et al. Phase I study of anti-VEGF monoclonal antibody bevacizumab and histone deacetylase inhibitor valproic acid in patients with advanced cancers. Cancer Chemother. Pharmacol. 2014, 73, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathkopf, D.E.; Picus, J.; Hussain, A.; Ellard, S.; Chi, K.N.; Nydam, T.; Allen-Freda, E.; Mishra, K.K.; Porro, M.G.; Scher, H.I.; et al. A phase 2 study of intravenous panobinostat in patients with castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2013, 72, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, A.C.; Alumkal, J.J.; Stein, M.N.; Taplin, M.E.; Babb, J.; Barnett, E.S.; Gomez-Pinillos, A.; Liu, X.; Moore, D.; DiPaola, R.; et al. Epigenetic Therapy with Panobinostat Combined with Bicalutamide Rechallenge in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 52–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathkopf, D.; Wong, B.Y.; Ross, R.W.; Anand, A.; Tanaka, E.; Woo, M.M.; Hu, J.; Dzik-Jurasz, A.; Yang, W.; Scher, H.I. A phase I study of oral panobinostat alone and in combination with docetaxel in patients with castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2010, 66, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.C.; Stein, M.N.; Alumkal, J.J.; Gomez-Pinillos, A.; Catamero, D.D.; Mayer, T.M.; Collins, F.; Beer, T.M.; DiPaola, R.S. A phase I/II randomized study of panobinostat and bicalutamide in castration-resistant prostate cancer (CRPC) patients progressing on second-line hormone therapy. J. Clin. Oncol. 2011, 29, 156. [Google Scholar] [CrossRef]
- Molife, L.R.; Attard, G.; Fong, P.C.; Karavasilis, V.; Reid, A.H.; Patterson, S.; Riggs, C.E., Jr.; Higano, C.; Stadler, W.M.; McCulloch, W.; et al. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC). Ann. Oncol. 2010, 21, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Eigl, B.J.; North, S.; Winquist, E.; Finch, D.; Wood, L.; Sridhar, S.S.; Powers, J.; Good, J.; Sharma, M.; Squire, J.A.; et al. A phase II study of the HDAC inhibitor SB939 in patients with castration resistant prostate cancer: NCIC clinical trials group study IND195. Investig. New Drugs 2015, 33, 969–976. [Google Scholar] [CrossRef]
- Ryan, Q.C.; Headlee, D.; Acharya, M.; Sparreboom, A.; Trepel, J.B.; Ye, J.; Figg, W.D.; Hwang, K.; Chung, E.J.; Murgo, A.; et al. Phase I and pharmacokinetic study of MS-275, a histone deacetylase inhibitor, in patients with advanced and refractory solid tumors or lymphoma. J. Clin. Oncol. 2005, 23, 3912–3922. [Google Scholar] [CrossRef]
- Varambally, S.; Yu, J.; Laxman, B.; Rhodes, D.R.; Mehra, R.; Tomlins, S.A.; Shah, R.B.; Chandran, U.; Monzon, F.A.; Becich, M.J.; et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 2005, 8, 393–406. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Hieronymus, H.; Schultz, N.; Gopalan, A.; Carver, B.S.; Chang, M.T.; Xiao, Y.; Heguy, A.; Huberman, K.; Bernstein, M.; Assel, M.; et al. Copy number alteration burden predicts prostate cancer relapse. Proc. Natl. Acad. Sci. USA 2014, 111, 11139–11144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Stelloo, S.; Nevedomskaya, E.; Kim, Y.; Schuurman, K.; Valle-Encinas, E.; Lobo, J.; Krijgsman, O.; Peeper, D.S.; Chang, S.L.; Feng, F.Y.; et al. Integrative epigenetic taxonomy of primary prostate cancer. Nat. Commun. 2018, 9, 4900. [Google Scholar] [CrossRef] [PubMed]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. Publisher Correction: The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2019, 51, 1194. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Wu, Y.P.; Ke, Z.B.; Liang, Y.C.; Cai, H.; Su, W.T.; Tao, X.; Chen, S.H.; Zheng, Q.S.; Wei, Y.; et al. Identification of key DNA methylation-driven genes in prostate adenocarcinoma: An integrative analysis of TCGA methylation data. J. Transl. Med. 2019, 17, 311. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.D.; Lin, N.; Lin, T.T.; Wu, Y.P.; Huang, P.; Ke, Z.B.; Lin, Y.Z.; Chen, S.H.; Zheng, Q.S.; Wei, Y.; et al. Identification of marker genes and cell subtypes in castration-resistant prostate cancer cells. J. Cancer 2021, 12, 1249–1257. [Google Scholar] [CrossRef] [PubMed]
- Martignano, F.; Gurioli, G.; Salvi, S.; Calistri, D.; Costantini, M.; Gunelli, R.; De Giorgi, U.; Foca, F.; Casadio, V. GSTP1 Methylation and Protein Expression in Prostate Cancer: Diagnostic Implications. Dis. Markers 2016, 2016, 4358292. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Freije, D.; Nusskern, D.R.; Okami, K.; Cairns, P.; Sidransky, D.; Isaacs, W.B.; Bova, G.S. Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic prostate cancer tissues. Cancer Res. 1998, 58, 204–209. [Google Scholar]
- Jarrard, D.F.; Bova, G.S.; Ewing, C.M.; Pin, S.S.; Nguyen, S.H.; Baylin, S.B.; Cairns, P.; Sidransky, D.; Herman, J.G.; Isaacs, W.B. Deletional, mutational, and methylation analyses of CDKN2 (p16/MTS1) in primary and metastatic prostate cancer. Genes Chromosomes Cancer 1997, 19, 90–96. [Google Scholar] [CrossRef]
- Ruggero, K.; Farran-Matas, S.; Martinez-Tebar, A.; Aytes, A. Epigenetic Regulation in Prostate Cancer Progression. Curr. Mol. Biol. Rep. 2018, 4, 101–115. [Google Scholar] [CrossRef]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat Rev Genet 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Uysal, F.; Cinar, O.; Can, A. Knockdown of Dnmt1 and Dnmt3a gene expression disrupts preimplantation embryo development through global DNA methylation. J. Assist. Reprod. Genet. 2021, 38, 3135–3144. [Google Scholar] [CrossRef]
- Turnham, D.J.; Bullock, N.; Dass, M.S.; Staffurth, J.N.; Pearson, H.B. The PTEN Conundrum: How to Target PTEN-Deficient Prostate Cancer. Cells 2020, 9, 2342. [Google Scholar] [CrossRef]
- Zhao, R.; Choi, B.Y.; Lee, M.H.; Bode, A.M.; Dong, Z. Implications of Genetic and Epigenetic Alterations of CDKN2A (p16(INK4a)) in Cancer. EBioMedicine 2016, 8, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, T.; Watanabe, M.; Suzuki, H.; Toyota, M.; Sekita, N.; Hirokawa, Y.; Mizokami, A.; Ito, H.; Yatani, R.; Shiraishi, T. Epigenetic regulation of androgen receptor gene expression in human prostate cancers. Lab. Investig. 2000, 80, 1789–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asrani, K.; Torres, A.F.C.; Woo, J.; Vidotto, T.; Tsai, H.K.; Luo, J.; Corey, E.; Hanratty, B.; Coleman, I.; Yegnasubramanian, S.; et al. Reciprocal YAP1 loss and INSM1 expression in neuroendocrine prostate cancer. J. Pathol. 2021, 255, 425–437. [Google Scholar] [CrossRef]
- Wang, L.; Ren, G.; Lin, B. Expression of 5-methylcytosine regulators is highly associated with the clinical phenotypes of prostate cancer and DNMTs expression predicts biochemical recurrence. Cancer Med. 2021, 10, 5681–5695. [Google Scholar] [CrossRef]
- Zhu, A.; Hopkins, K.M.; Friedman, R.A.; Bernstock, J.D.; Broustas, C.G.; Lieberman, H.B. DNMT1 and DNMT3B regulate tumorigenicity of human prostate cancer cells by controlling RAD9 expression through targeted methylation. Carcinogenesis 2021, 42, 220–231. [Google Scholar] [CrossRef]
- Mancini, M.; Grasso, M.; Muccillo, L.; Babbio, F.; Precazzini, F.; Castiglioni, I.; Zanetti, V.; Rizzo, F.; Pistore, C.; De Marino, M.G.; et al. DNMT3A epigenetically regulates key microRNAs involved in epithelial-to-mesenchymal transition in prostate cancer. Carcinogenesis 2021, 42, 1449–1460. [Google Scholar] [CrossRef]
- Lin, J.; Haffner, M.C.; Zhang, Y.; Lee, B.H.; Brennen, W.N.; Britton, J.; Kachhap, S.K.; Shim, J.S.; Liu, J.O.; Nelson, W.G.; et al. Disulfiram is a DNA demethylating agent and inhibits prostate cancer cell growth. Prostate 2011, 71, 333–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffey, K.; Rogerson, L.; Ryan-Munden, C.; Alkharaif, D.; Stockley, J.; Heer, R.; Sahadevan, K.; O’Neill, D.; Jones, D.; Darby, S.; et al. The lysine demethylase, KDM4B, is a key molecule in androgen receptor signalling and turnover. Nucleic Acids Res. 2013, 41, 4433–4446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Wang, C.; Yang, J.; Guo, Y.; Wu, Y.; Li, X. Metformin inhibits SUV39H1-mediated migration of prostate cancer cells. Oncogenesis 2017, 6, e324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Askew, E.B.; Bai, S.; Parris, A.B.; Minges, J.T.; Wilson, E.M. Androgen receptor regulation by histone methyltransferase Suppressor of variegation 3-9 homolog 2 and Melanoma antigen-A11. Mol. Cell Endocrinol. 2017, 443, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Xu, A.M. SET and MYND domain containing protein 3 in cancer. Am. J. Transl. Res. 2017, 9, 1–14. [Google Scholar]
- Li, Q.; Li, Y.; Wang, Y.; Cui, Z.; Gong, L.; Qu, Z.; Zhong, Y.; Zhou, J.; Zhou, Y.; Gao, Y.; et al. Quantitative proteomic study of human prostate cancer cells with different metastatic potentials. Int. J. Oncol. 2016, 48, 1437–1446. [Google Scholar] [CrossRef] [Green Version]
- Stopa, N.; Krebs, J.E.; Shechter, D. The PRMT5 arginine methyltransferase: Many roles in development, cancer and beyond. Cell Mol. Life Sci. 2015, 72, 2041–2059. [Google Scholar] [CrossRef]
- Grypari, I.M.; Logotheti, S.; Zolota, V.; Troncoso, P.; Efstathiou, E.; Bravou, V.; Melachrinou, M.; Logothetis, C.; Tzelepi, V. The protein arginine methyltransferases (PRMTs) PRMT1 and CARM1 as candidate epigenetic drivers in prostate cancer progression. Medicine 2021, 100, e27094. [Google Scholar] [CrossRef]
- Keats, J.J.; Maxwell, C.A.; Taylor, B.J.; Hendzel, M.J.; Chesi, M.; Bergsagel, P.L.; Larratt, L.M.; Mant, M.J.; Reiman, T.; Belch, A.R.; et al. Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16;q32)-positive multiple myeloma patients. Blood 2005, 105, 4060–4069. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Xue, W.; Yuan, H.; Dong, B.; Ding, Y.; Liu, Y.; Jiang, M.; Kan, S.; Sun, T.; Ren, J.; et al. AKT-mediated stabilization of histone methyltransferase WHSC1 promotes prostate cancer metastasis. J. Clin. Investig. 2017, 127, 1284–1302. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Trojer, P.; Xu, C.F.; Cheung, P.; Kuo, A.; Drury, W.J., 3rd; Qiao, Q.; Neubert, T.A.; Xu, R.M.; Gozani, O.; et al. The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J. Biol. Chem. 2009, 284, 34283–34295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezponda, T.; Popovic, R.; Shah, M.Y.; Martinez-Garcia, E.; Zheng, Y.; Min, D.J.; Will, C.; Neri, A.; Kelleher, N.L.; Yu, J.; et al. The histone methyltransferase MMSET/WHSC1 activates TWIST1 to promote an epithelial-mesenchymal transition and invasive properties of prostate cancer. Oncogene 2013, 32, 2882–2890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.; Guo, L.; Duan, Z.J.; Tepper, C.G.; Xue, L.; Chen, X.; Kung, H.J.; Gao, A.C.; Zou, J.X.; Chen, H.W. Histone methyltransferase NSD2/MMSET mediates constitutive NF-κB signaling for cancer cell proliferation, survival, and tumor growth via a feed-forward loop. Mol. Cell Biol. 2012, 32, 3121–3131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.B.; Choi, Y.; Lee, J.M.; Choi, K.C.; Kim, H.C.; Yoo, J.Y.; Lee, Y.H.; Yoon, H.G. The histone methyltransferase, NSD2, enhances androgen receptor-mediated transcription. FEBS Lett. 2009, 583, 1880–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melling, N.; Thomsen, E.; Tsourlakis, M.C.; Kluth, M.; Hube-Magg, C.; Minner, S.; Koop, C.; Graefen, M.; Heinzer, H.; Wittmer, C.; et al. Overexpression of enhancer of zeste homolog 2 (EZH2) characterizes an aggressive subset of prostate cancers and predicts patient prognosis independently from pre- and postoperatively assessed clinicopathological parameters. Carcinogenesis 2015, 36, 1333–1340. [Google Scholar] [CrossRef] [Green Version]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbe, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Lee, Y.; Lu, X.; Song, B.; Fong, K.W.; Cao, Q.; Licht, J.D.; Zhao, J.C.; Yu, J. Polycomb- and Methylation-Independent Roles of EZH2 as a Transcription Activator. Cell Rep. 2018, 25, 2808–2820.e4. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Tien, J.C.; Vo, J.; Tan, M.; Parolia, A.; Zhang, Y.; Wang, L.; Qiao, Y.; Shukla, S.; Wang, X.; et al. Epigenetic Reprogramming with Antisense Oligonucleotides Enhances the Effectiveness of Androgen Receptor Inhibition in Castration-Resistant Prostate Cancer. Cancer Res. 2018, 78, 5731–5740. [Google Scholar] [CrossRef] [Green Version]
- Morel, K.L.; Sheahan, A.V.; Burkhart, D.L.; Baca, S.C.; Boufaied, N.; Liu, Y.; Qiu, X.; Canadas, I.; Roehle, K.; Heckler, M.; et al. EZH2 inhibition activates a dsRNA-STING-interferon stress axis that potentiates response to PD-1 checkpoint blockade in prostate cancer. Nat. Cancer 2021, 2, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Valdes-Mora, F.; Gould, C.M.; Colino-Sanguino, Y.; Qu, W.; Song, J.Z.; Taylor, K.M.; Buske, F.A.; Statham, A.L.; Nair, S.S.; Armstrong, N.J.; et al. Acetylated histone variant H2A.Z is involved in the activation of neo-enhancers in prostate cancer. Nat. Commun. 2017, 8, 1346. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Wang, C.; Reutens, A.T.; Wang, J.; Angeletti, R.H.; Siconolfi-Baez, L.; Ogryzko, V.; Avantaggiati, M.L.; Pestell, R.G. p300 and p300/cAMP-response element-binding protein-associated factor acetylate the androgen receptor at sites governing hormone-dependent transactivation. J. Biol. Chem. 2000, 275, 20853–20860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Garcia, J.; Chan, E.; de la Cruz, C.; Segal, E.; Merchant, M.; Kharbanda, S.; Raisner, R.; Haverty, P.M.; Modrusan, Z.; et al. Therapeutic Targeting of the CBP/p300 Bromodomain Blocks the Growth of Castration-Resistant Prostate Cancer. Cancer Res. 2017, 77, 5564–5575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; He, D.; Cheng, L.; Huang, C.; Zhang, Y.; Rao, X.; Kong, Y.; Li, C.; Zhang, Z.; Liu, J.; et al. p300/CBP inhibition enhances the efficacy of programmed death-ligand 1 blockade treatment in prostate cancer. Oncogene 2020, 39, 3939–3951. [Google Scholar] [CrossRef]
- Wu, C.; Jin, X.; Yang, J.; Yang, Y.; He, Y.; Ding, L.; Pan, Y.; Chen, S.; Jiang, J.; Huang, H. Inhibition of EZH2 by chemo- and radiotherapy agents and small molecule inhibitors induces cell death in castration-resistant prostate cancer. Oncotarget 2016, 7, 3440–3452. [Google Scholar] [CrossRef]
- Ge, R.; Wang, Z.; Montironi, R.; Jiang, Z.; Cheng, M.; Santoni, M.; Huang, K.; Massari, F.; Lu, X.; Cimadamore, A.; et al. Epigenetic modulations and lineage plasticity in advanced prostate cancer. Ann. Oncol. 2020, 31, 470–479. [Google Scholar] [CrossRef] [Green Version]
- Davies, A.; Zoubeidi, A.; Selth, L.A. The epigenetic and transcriptional landscape of neuroendocrine prostate cancer. Endocr. Relat. Cancer 2020, 27, R35–R50. [Google Scholar] [CrossRef]
- Welti, J.; Sharp, A.; Brooks, N.; Yuan, W.; McNair, C.; Chand, S.N.; Pal, A.; Figueiredo, I.; Riisnaes, R.; Gurel, B.; et al. Targeting the p300/CBP Axis in Lethal Prostate Cancer. Cancer Discov. 2021, 11, 1118–1137. [Google Scholar] [CrossRef]
- Urbanucci, A.; Mills, I.G. Bromodomain-containing proteins in prostate cancer. Mol. Cell Endocrinol. 2018, 462, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devaiah, B.N.; Case-Borden, C.; Gegonne, A.; Hsu, C.H.; Chen, Q.; Meerzaman, D.; Dey, A.; Ozato, K.; Singer, D.S. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat. Struct. Mol. Biol. 2016, 23, 540–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Faivre, E.J.; Wilcox, D.; Lin, X.; Hessler, P.; Torrent, M.; He, W.; Uziel, T.; Albert, D.H.; McDaniel, K.; Kati, W.; et al. Exploitation of Castration-Resistant Prostate Cancer Transcription Factor Dependencies by the Novel BET Inhibitor ABBV-075. Mol. Cancer Res. 2017, 15, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Coleman, D.J.; Gao, L.; King, C.J.; Schwartzman, J.; Urrutia, J.; Sehrawat, A.; Tayou, J.; Balter, A.; Burchard, J.; Chiotti, K.E.; et al. BET bromodomain inhibition blocks the function of a critical AR-independent master regulator network in lethal prostate cancer. Oncogene 2019, 38, 5658–5669. [Google Scholar] [CrossRef] [PubMed]
- Groner, A.C.; Cato, L.; de Tribolet-Hardy, J.; Bernasocchi, T.; Janouskova, H.; Melchers, D.; Houtman, R.; Cato, A.C.B.; Tschopp, P.; Gu, L.; et al. TRIM24 Is an Oncogenic Transcriptional Activator in Prostate Cancer. Cancer Cell 2016, 29, 846–858. [Google Scholar] [CrossRef] [Green Version]
- Peña-Hernández, R.; Aprigliano, R.; Carina Frommel, S.; Pietrzak, K.; Steiger, S.; Roganowicz, M.; Lerra, L.; Bizzarro, J.; Santoro, R. BAZ2A-mediated repression via H3K14ac-marked enhancers promotes prostate cancer stem cells. EMBO Rep. 2021, 22, e53014. [Google Scholar] [CrossRef]
- Zhao, D.; Lu, X.; Wang, G.; Lan, Z.; Liao, W.; Li, J.; Liang, X.; Chen, J.R.; Shah, S.; Shang, X.; et al. Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN-deficient cancer. Nature 2017, 542, 484–488. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Salvia, M.; Esteller, M. Bromodomain inhibitors and cancer therapy: From structures to applications. Epigenetics 2017, 12, 323–339. [Google Scholar] [CrossRef]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef] [Green Version]
- Pakneshan, P.; Xing, R.H.; Rabbani, S.A. Methylation status of uPA promoter as a molecular mechanism regulating prostate cancer invasion and growth in vitro and in vivo. FASEB J. 2003, 17, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, J.; Zhang, K.; Lu, G.; Liu, Y.; Ren, K.; Wang, W.; Xin, D.; Xu, L.; Mao, H.; et al. Ten-eleven translocation 1 mediated-DNA hydroxymethylation is required for myelination and remyelination in the mouse brain. Nat. Commun. 2021, 12, 5091. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, M.L.; Das, S.; Im, K.M.; Turan, S.; Berndt, S.I.; Li, H.; Lou, H.; Brodie, S.A.; Billaud, J.N.; Zhang, T.; et al. TET2 binds the androgen receptor and loss is associated with prostate cancer. Oncogene 2017, 36, 2172–2183. [Google Scholar] [CrossRef] [Green Version]
- Takayama, K.; Misawa, A.; Suzuki, T.; Takagi, K.; Hayashizaki, Y.; Fujimura, T.; Homma, Y.; Takahashi, S.; Urano, T.; Inoue, S. TET2 repression by androgen hormone regulates global hydroxymethylation status and prostate cancer progression. Nat. Commun. 2015, 6, 8219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.H.; Peng, K.L.; Kang, M.L.; Chen, Y.R.; Yang, Y.C.; Tsai, C.H.; Chu, C.S.; Jeng, Y.M.; Chen, Y.T.; Lin, F.M.; et al. TET1 suppresses cancer invasion by activating the tissue inhibitors of metalloproteinases. Cell Rep. 2012, 2, 568–579. [Google Scholar] [CrossRef] [Green Version]
- Metzger, E.; Wissmann, M.; Yin, N.; Muller, J.M.; Schneider, R.; Peters, A.H.; Gunther, T.; Buettner, R.; Schule, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef]
- Hayami, S.; Kelly, J.D.; Cho, H.S.; Yoshimatsu, M.; Unoki, M.; Tsunoda, T.; Field, H.I.; Neal, D.E.; Yamaue, H.; Ponder, B.A.; et al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int. J. Cancer 2011, 128, 574–586. [Google Scholar] [CrossRef]
- Kahl, P.; Gullotti, L.; Heukamp, L.C.; Wolf, S.; Friedrichs, N.; Vorreuther, R.; Solleder, G.; Bastian, P.J.; Ellinger, J.; Metzger, E.; et al. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006, 66, 11341–11347. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; He, H.H.; Chen, S.; Coleman, I.; Wang, H.; Fang, Z.; Chen, S.; Nelson, P.S.; Liu, X.S.; Brown, M.; et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell 2011, 20, 457–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.; Chen, S.; Han, D.; Wang, Z.; Li, M.; Han, W.; Besschetnova, A.; Liu, M.; Zhou, F.; Barrett, D.; et al. Chromatin binding of FOXA1 is promoted by LSD1-mediated demethylation in prostate cancer. Nat. Genet. 2020, 52, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Ahmed, M.; Guo, H.; Soares, F.; Hua, J.T.; Gao, S.; Lu, C.; Poon, C.; Han, W.; Langstein, J.; et al. LSD1-Mediated Epigenetic Reprogramming Drives CENPE Expression and Prostate Cancer Progression. Cancer Res. 2017, 77, 5479–5490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehrawat, A.; Gao, L.; Wang, Y.; Bankhead, A., 3rd; McWeeney, S.K.; King, C.J.; Schwartzman, J.; Urrutia, J.; Bisson, W.H.; Coleman, D.J.; et al. LSD1 activates a lethal prostate cancer gene network independently of its demethylase function. Proc. Natl. Acad. Sci. USA 2018, 115, E4179–E4188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Dhar, S.S.; Chen, T.Y.; Kan, P.Y.; Wei, Y.; Kim, J.H.; Chan, C.H.; Lin, H.K.; Hung, M.C.; Lee, M.G. JARID1D Is a Suppressor and Prognostic Marker of Prostate Cancer Invasion and Metastasis. Cancer Res. 2016, 76, 831–843. [Google Scholar] [CrossRef] [Green Version]
- Komura, K.; Jeong, S.H.; Hinohara, K.; Qu, F.; Wang, X.; Hiraki, M.; Azuma, H.; Lee, G.S.; Kantoff, P.W.; Sweeney, C.J. Resistance to docetaxel in prostate cancer is associated with androgen receptor activation and loss of KDM5D expression. Proc. Natl. Acad. Sci. USA 2016, 113, 6259–6264. [Google Scholar] [CrossRef] [Green Version]
- Stein, J.; Majores, M.; Rohde, M.; Lim, S.; Schneider, S.; Krappe, E.; Ellinger, J.; Dietel, M.; Stephan, C.; Jung, K.; et al. KDM5C is overexpressed in prostate cancer and is a prognostic marker for prostate-specific antigen-relapse following radical prostatectomy. Am. J. Pathol. 2014, 184, 2430–2437. [Google Scholar] [CrossRef]
- Tang, D.; He, J.; Dai, Y.; Zhou, H.; Zhang, C.; Leng, Q.; Geng, X.; Fu, D.; Jiang, H.; Sun, R.; et al. Targeting KDM6A Suppresses SREBP1c-Dependent Lipid Metabolism and Prostate Tumorigenesis. Cancer Res 2021. [Google Scholar] [CrossRef]
- Yildirim-Buharalioglu, G. KDM6B Regulates Prostate Cancer Cell Proliferation by Controlling c-MYC Expression. Mol. Pharmacol. 2021, 101, 106–119. [Google Scholar] [CrossRef]
- Xiang, Y.; Zhu, Z.; Han, G.; Ye, X.; Xu, B.; Peng, Z.; Ma, Y.; Yu, Y.; Lin, H.; Chen, A.P.; et al. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 19226–19231. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Janknecht, R. Activation of androgen receptor by histone demethylases JMJD2A and JMJD2D. Biochem. Biophys. Res. Commun. 2007, 359, 742–746. [Google Scholar] [CrossRef] [PubMed]
- Wissmann, M.; Yin, N.; Muller, J.M.; Greschik, H.; Fodor, B.D.; Jenuwein, T.; Vogler, C.; Schneider, R.; Gunther, T.; Buettner, R.; et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol. 2007, 9, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Yamane, K.; Toumazou, C.; Tsukada, Y.; Erdjument-Bromage, H.; Tempst, P.; Wong, J.; Zhang, Y. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 2006, 125, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, L.; Chen, Z.; Lu, J.; Liang, Y.; Wang, M.; Roggero, C.M.; Zhang, Q.J.; Gao, J.; Fang, Y.; Cao, J.; et al. Histone lysine demethylase KDM4B regulates the alternative splicing of the androgen receptor in response to androgen deprivation. Nucleic Acids Res. 2019, 47, 11623–11636. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Rai, G.; Roggero, C.; Zhang, Q.J.; Wei, Q.; Ma, S.H.; Zhou, Y.; Santoyo, J.; Martinez, E.D.; Xiao, G.; et al. KDM4/JMJD2 Histone Demethylase Inhibitors Block Prostate Tumor Growth by Suppressing the Expression of AR and BMYB-Regulated Genes. Chem. Biol. 2015, 22, 1185–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarac, H.; Morova, T.; Pires, E.; McCullagh, J.; Kaplan, A.; Cingoz, A.; Bagci-Onder, T.; Onder, T.; Kawamura, A.; Lack, N.A. Systematic characterization of chromatin modifying enzymes identifies KDM3B as a critical regulator in castration resistant prostate cancer. Oncogene 2020, 39, 2187–2201. [Google Scholar] [CrossRef] [Green Version]
- Maina, P.K.; Shao, P.; Liu, Q.; Fazli, L.; Tyler, S.; Nasir, M.; Dong, X.; Qi, H.H. c-MYC drives histone demethylase PHF8 during neuroendocrine differentiation and in castration-resistant prostate cancer. Oncotarget 2016, 7, 75585–75602. [Google Scholar] [CrossRef] [Green Version]
- Tong, D.; Liu, Q.; Liu, G.; Yuan, W.; Wang, L.; Guo, Y.; Lan, W.; Zhang, D.; Dong, S.; Wang, Y.; et al. The HIF/PHF8/AR axis promotes prostate cancer progression. Oncogenesis 2016, 5, e283. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.J.; Pochampalli, M.; Wang, L.Y.; Zou, J.X.; Li, P.S.; Hsu, S.C.; Wang, B.J.; Huang, S.H.; Yang, P.; Yang, J.C.; et al. KDM8/JMJD5 as a dual coactivator of AR and PKM2 integrates AR/EZH2 network and tumor metabolism in CRPC. Oncogene 2019, 38, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Civenni, G.; Zoppi, G.; Vazquez, R.; Shinde, D.; Paganoni, A.; Kokanovic, A.; Lee, S.H.; Ruggeri, B.; Carbone, G.M.; Catapano, C.V. Abstract 1379: INCB059872, a novel FAD-directed LSD1 Inhibitor, is active in prostate cancer models and impacts prostate cancer stem-like cells. Cancer Res. 2018, 78, 1379. [Google Scholar] [CrossRef]
- Lavery, D.N.; Bevan, C.L. Androgen receptor signalling in prostate cancer: The functional consequences of acetylation. J. Biomed. Biotechnol. 2011, 2011, 862125. [Google Scholar] [CrossRef] [Green Version]
- Ruscetti, M.; Dadashian, E.L.; Guo, W.; Quach, B.; Mulholland, D.J.; Park, J.W.; Tran, L.M.; Kobayashi, N.; Bianchi-Frias, D.; Xing, Y.; et al. HDAC inhibition impedes epithelial-mesenchymal plasticity and suppresses metastatic, castration-resistant prostate cancer. Oncogene 2016, 35, 3781–3795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakshminarasimhan, R.; Liang, G. The Role of DNA Methylation in Cancer. Adv. Exp. Med. Biol. 2016, 945, 151–172. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.K.; De Carvalho, D.D.; Jones, P.A. Epigenetic modifications as therapeutic targets. Nat. Biotechnol. 2010, 28, 1069–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juo, Y.Y.; Gong, X.J.; Mishra, A.; Cui, X.; Baylin, S.B.; Azad, N.S.; Ahuja, N. Epigenetic therapy for solid tumors: From bench science to clinical trials. Epigenomics 2015, 7, 215–235. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, J.; Issa, J.P. Epigenetic aspects of MDS and its molecular targeted therapy. Int. J. Hematol. 2013, 97, 175–182. [Google Scholar] [CrossRef] [Green Version]
- Graça, I.; Pereira-Silva, E.; Henrique, R.; Packham, G.; Crabb, S.J.; Jerónimo, C. Epigenetic modulators as therapeutic targets in prostate cancer. Clin. Epigenet. 2016, 8, 98. [Google Scholar] [CrossRef] [Green Version]
- Helm, M.; Motorin, Y. Detecting RNA modifications in the epitranscriptome: Predict and validate. Nat. Rev. Genet. 2017, 18, 275–291. [Google Scholar] [CrossRef]
- Natchiar, S.K.; Myasnikov, A.G.; Kratzat, H.; Hazemann, I.; Klaholz, B.P. Visualization of chemical modifications in the human 80S ribosome structure. Nature 2017, 551, 472–477. [Google Scholar] [CrossRef]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Pitkowski, P.; Bagiski, B.; Wirecki, T.K.; de Crcy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2017, 46, D303–D307. [Google Scholar] [CrossRef]
- Garcia-Vilchez, R.; Sevilla, A.; Blanco, S. Post-transcriptional regulation by cytosine-5 methylation of RNA. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1862, 240–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krutyhołowa, R.; Zakrzewski, K.; Glatt, S. Charging the code—tRNA modification complexes. Curr. Opin. Struct. Biol. 2019, 55, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Pan, T. Modifications and functional genomics of human transfer RNA. Cell Res. 2018, 28, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Sloan, K.E.; Warda, A.S.; Sharma, S.; Entian, K.-D.; Lafontaine, D.; Bohnsack, M.T. Tuning the ribosome: The influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol. 2017, 14, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Nombela, P.; Miguel-López, B.; Blanco, S. The role of m(6)A, m(5)C and Ψ RNA modifications in cancer: Novel therapeutic opportunities. Mol. Cancer 2021, 20, 18. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P. CpG-rich islands and the function of DNA methylation. Nature 1986, 321, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Blanco, S.; Dietmann, S.; Flores, J.V.; Hussain, S.; Kutter, C.; Humphreys, P.; Lukk, M.; Lombard, P.; Treps, L.; Popis, M.; et al. Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J. 2014, 33, 2020–2039. [Google Scholar] [CrossRef]
- Li, Y.; Ge, Y.-Z.; Xu, L.; Xu, Z.; Dou, Q.; Jia, R. The Potential Roles of RNA N6-Methyladenosine in Urological Tumors. Front. Cell Dev. Biol. 2020, 8, 579919. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Zhou, Z.; Lv, J.; Yu, H.; Han, J.; Yang, X.; Feng, D.; Wu, Q.; Yuan, B.; Lu, Q.; Yang, H. Mechanism of RNA modification N6-methyladenosine in human cancer. Mol. Cancer 2020, 19, 104. [Google Scholar] [CrossRef]
- Warda, A.S.; Kretschmer, J.; Hackert, P.; Lenz, C.; Urlaub, H.; Höbartner, C.; Sloan, K.E.; Bohnsack, M.T. Human METTL16 is a N(6)-methyladenosine (m(6)A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 2017, 18, 2004–2014. [Google Scholar] [CrossRef] [PubMed]
- van Tran, N.; Ernst, F.G.M.; Hawley, B.R.; Zorbas, C.; Ulryck, N.; Hackert, P.; Bohnsack, K.E.; Bohnsack, M.T.; Jaffrey, S.R.; Graille, M.; et al. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 2019, 47, 7719–7733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell. 2017, 31, 127–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.J.; Chen, Q.; et al. Reversible methylation of m(6)Am in the 5’ cap controls mRNA stability. Nature 2017, 541, 371–375. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Liu, Y.; Liu, J.; Xu, T.; Cheng, G.; Shou, Y.; Tong, J.; Liu, L.; Zhou, L.; Xiao, W.; et al. The Identification of Critical m6A RNA Methylation Regulators as Malignant Prognosis Factors in Prostate Adenocarcinoma. Front. Genet. 2020, 11, 602485. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Xie, X.; Huang, Y.; Meng, S.; Li, Y.; Wang, H.; Hu, Y. N6-methyladenosine RNA methylation regulators contribute to the progression of prostate cancer. J. Cancer 2021, 12, 682–692. [Google Scholar] [CrossRef]
- Ji, G.; Huang, C.; He, S.; Gong, Y.; Song, G.; Li, X.; Zhou, L. Comprehensive analysis of m6A regulators prognostic value in prostate cancer. Aging 2020, 12, 14863–14884. [Google Scholar] [CrossRef]
- Zhang, Q.; Luan, J.; Song, L.; Wei, X.; Xia, J.; Song, N. Malignant Evaluation and Clinical Prognostic Values of M6A RNA Methylation Regulators in Prostate Cancer. J. Cancer 2021, 12, 3575–3586. [Google Scholar] [CrossRef]
- Su, H.; Wang, Y.; Li, H. RNA m6A Methylation Regulators Multi-Omics Analysis in Prostate Cancer. Front. Genet. 2021, 12, 768041. [Google Scholar] [CrossRef]
- Cai, J.; Yang, F.; Zhan, H.; Situ, J.; Li, W.; Mao, Y.; Luo, Y. RNA m(6)A Methyltransferase METTL3 Promotes the Growth of Prostate Cancer by Regulating Hedgehog Pathway. Onco. Targets Ther. 2019, 12, 9143–9152. [Google Scholar] [CrossRef] [Green Version]
- Li, E.; Wei, B.; Wang, X.; Kang, R. METTL3 enhances cell adhesion through stabilizing integrin β1 mRNA via an m6A-HuR-dependent mechanism in prostatic carcinoma. Am. J. Cancer. Res. 2020, 10, 1012–1025. [Google Scholar] [PubMed]
- Yuan, Y.; Du, Y.; Wang, L.; Liu, X. The M6A methyltransferase METTL3 promotes the development and progression of prostate carcinoma via mediating MYC methylation. J. Cancer. 2020, 11, 3588–3595. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Pan, C.; Wang, X.; Xu, D.; Ma, Y.; Hu, J.; Chen, P.; Xiang, Z.; Rao, Q.; Han, X. Silencing of METTL3 effectively hinders invasion and metastasis of prostate cancer cells. Theranostics 2021, 11, 7640–7657. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Wei, Y.; Zen, C.; Xiong, W.; Niu, Y.; Zhao, Y. Long non-coding RNA NEAT1 promotes bone metastasis of prostate cancer through N6-methyladenosine. Mol. Cancer 2020, 19, 171. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.; Yin, C.; Lin, K.; Li, Y.; Yang, Q.; Wu, Z.; Du, H.; Ren, D.; Dai, Y.; Peng, X. m 6 A modification of lncRNA PCAT6 promotes bone metastasis in prostate cancer through IGF2BP2-mediated IGF1R mRNA stabilization. Clin. Transl. Med. 2021, 11, e426. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, H.; Zheng, J.; Shao, C. Analysis of RNA m 6 A methylation regulators and tumour immune cell infiltration characterization in prostate cancer. Artif. Cells Nanomed. Biotechnol. 2021, 49, 407–435. [Google Scholar] [CrossRef]
- Liu, Z.; Zhong, J.; Zeng, J.; Duan, X.; Lu, J.; Sun, X.; Liu, Q.; Liang, Y.; Lin, Z.; Zhong, W.; et al. Characterization of the m6A-Associated Tumor Immune Microenvironment in Prostate Cancer to Aid Immunotherapy. Front. Immunol. 2021, 12, 735170. [Google Scholar] [CrossRef]
- Barros-Silva, D.; Lobo, J.; Guimarães-Teixeira, C.; Carneiro, I.; Oliveira, J.; Martens-Uzunova, E.S.; Henrique, R.; Jerónimo, C. VIRMA-Dependent N6-Methyladenosine Modifications Regulate the Expression of Long Non-Coding RNAs CCAT1 and CCAT2 in Prostate Cancer. Cancers 2020, 12, 771. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Xie, H.; Ying, Y.; Chen, H.; Yan, H.; He, L.; Xu, M.; Xu, X.; Liang, Z.; Liu, B.; et al. YTHDF2 mediates the mRNA degradation of the tumor suppressors to induce AKT phosphorylation in N6-methyladenosine-dependent way in prostate cancer. Mol. Cancer 2020, 19, 152. [Google Scholar] [CrossRef]
- Du, C.; Lv, C.; Feng, Y.; Yu, S. Activation of the KDM5A/miRNA-495/YTHDF2/m6A-MOB3B axis facilitates prostate cancer progression. J. Exp. Clin. Cancer Res. 2020, 39, 223. [Google Scholar] [CrossRef]
- Zhu, K.; Li, Y.; Xu, Y. The FTO m6A demethylase inhibits the invasion and migration of prostate cancer cells by regulating total m6A levels. Life Sci. 2021, 271, 119180. [Google Scholar] [CrossRef] [PubMed]
- Dolbois, A.; Bedi, R.K.; Bochenkova, E.; Müller, A.; Moroz-Omori, E.V.; Huang, D.; Caflisch, A. 1,4,9-Triazaspiro[5.5]undecan-2-one Derivatives as Potent and Selective METTL3 Inhibitors. J. Med. Chem. 2021, 64, 12738–12760. [Google Scholar] [CrossRef] [PubMed]
- Yankova, E.; Blackaby, W.; Albertella, M.; Rak, J.; De Braekeleer, E.; Tsagkogeorga, G.; Pilka, E.S.; Aspris, D.; Leggate, D.; Hendrick, A.G.; et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 2021, 593, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Squires, J.E.; Patel, H.R.; Nousch, M.; Sibbritt, T.; Humphreys, D.T.; Parker, B.J.; Suter, C.M.; Preiss, T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012, 40, 5023–5033. [Google Scholar] [CrossRef] [PubMed]
- Motorin, Y.; Lyko, F.; Helm, M. 5-methylcytosine in RNA: Detection, enzymatic formation and biological functions. Nucleic Acids Res. 2010, 38, 1415–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bantis, A.; Giannopoulos, A.; Gonidi, M.; Liossi, A.; Aggelonidou, E.; Petrakakou, E.; Athanassiades, P.; Athanassiadou, P. Expression of p120, Ki-67 and PCNA as proliferation biomarkers in imprint smears of prostate carcinoma and their prognostic value. Cytopathology 2004, 15, 25–31. [Google Scholar] [CrossRef]
- Kallakury, B.V.; Sheehan, C.E.; Rhee, S.J.; Fisher, H.A.; Kaufman, R.P., Jr.; Rifkin, M.D.; Ross, J.S. The prognostic significance of proliferation-associated nucleolar protein p120 expression in prostate adenocarcinoma: A comparison with cyclins A and B1, Ki-67, proliferating cell nuclear antigen, and p34cdc2. Cancer 1999, 85, 1569–1576. [Google Scholar] [CrossRef]
- Sharma, S.; Yang, J.; Watzinger, P.; Kotter, P.; Entian, K.D. Yeast Nop2 and Rcm1 methylate C2870 and C2278 of the 25S rRNA, respectively. Nucleic Acids Res. 2013, 41, 9062–9076. [Google Scholar] [CrossRef] [Green Version]
- Kar, S.P.; Beesley, J.; Amin Al Olama, A.; Michailidou, K.; Tyrer, J.; Kote-Jarai, Z.; Lawrenson, K.; Lindstrom, S.; Ramus, S.J.; Thompson, D.J.; et al. Genome-Wide Meta-Analyses of Breast, Ovarian, and Prostate Cancer Association Studies Identify Multiple New Susceptibility Loci Shared by at Least Two Cancer Types. Cancer Discov. 2016, 6, 1052–1067. [Google Scholar] [CrossRef] [Green Version]
- Metodiev, M.D.; Spåhr, H.; Loguercio Polosa, P.; Meharg, C.; Becker, C.; Altmueller, J.; Habermann, B.; Larsson, N.-G.; Ruzzenente, B. NSUN4 is a dual function mitochondrial protein required for both methylation of 12S rRNA and coordination of mitoribosomal assembly. PLoS Genet. 2014, 10, e1004110. [Google Scholar] [CrossRef] [Green Version]
- Delatte, B.; Wang, F.; Ngoc, L.V.; Collignon, E.; Bonvin, E.; Deplus, R.; Calonne, E.; Hassabi, B.; Putmans, P.; Awe, S.; et al. RNA biochemistry. Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science 2016, 351, 282–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haag, S.; Sloan, K.E.; Ranjan, N.; Warda, A.S.; Kretschmer, J.; Blessing, C.; Hübner, B.; Seikowski, J.; Dennerlein, S.; Rehling, P.; et al. NSUN3 and ABH1 modify the wobble position of mt-tRNAMet to expand codon recognition in mitochondrial translation. EMBO J. 2016, 35, 2104–2119. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Guerrero, C.R.; Zhong, N.; Amato, N.J.; Liu, Y.; Liu, S.; Cai, Q.; Ji, D.; Jin, S.G.; Niedernhofer, L.J.; et al. Tet-mediated formation of 5-hydroxymethylcytosine in RNA. J. Am. Chem. Soc. 2014, 136, 11582–11585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamdar, S.; Isserlin, R.; Van der Kwast, T.; Zlotta, A.R.; Bader, G.D.; Fleshner, N.E.; Bapat, B. Exploring targets of TET2-mediated methylation reprogramming as potential discriminators of prostate cancer progression. Clin. Epigenet. 2019, 11, 54. [Google Scholar] [CrossRef] [Green Version]
- Delaunay, S.; Frye, M. RNA modifications regulating cell fate in cancer. Nat. Cell Biol. 2019, 21, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, I.; Tzelepis, K.; Pandolfini, L.; Shi, J.; Millán-Zambrano, G.; Robson, S.C.; Aspris, D.; Migliori, V.; Bannister, A.J.; Han, N.; et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature 2017, 552, 126–131. [Google Scholar] [CrossRef]
- Blanco, S.; Bandiera, R.; Popis, M.; Hussain, S.; Lombard, P.; Aleksic, J.; Sajini, A.; Tanna, H.; Cortes-Garrido, R.; Gkatza, N.; et al. Stem cell function and stress response are controlled by protein synthesis. Nature 2016, 534, 335–340. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, M.; Fujiwara, M.; Hori, M.; Okada, K.; Yazama, F.; Konishi, H.; Xiao, Y.; Qi, G.; Shimamoto, F.; Ota, T.; et al. tRNA modifying enzymes, NSUN2 and METTL1, determine sensitivity to 5-fluorouracil in HeLa cells. PLoS Genet. 2014, 10, e1004639. [Google Scholar] [CrossRef]
- Rintala-Dempsey, A.C.; Kothe, U. Eukaryotic stand-alone pseudouridine synthases—RNA modifying enzymes and emerging regulators of gene expression? RNA Biol. 2017, 14, 1185–1196. [Google Scholar] [CrossRef] [Green Version]
- Stockert, J.A.; Weil, R.; Yadav, K.K.; Kyprianou, N.; Tewari, A.K. Pseudouridine as a novel biomarker in prostate cancer. Urol. Oncol. 2021, 39, 63–71. [Google Scholar] [CrossRef]
- Yu, Y.T.; Meier, U.T. RNA-guided isomerization of uridine to pseudouridine–Pseudouridylation. RNA Biol. 2014, 11, 1483–1494. [Google Scholar] [CrossRef] [Green Version]
- Levi, O.; Arava, Y.S. Pseudouridine-mediated translation control of mRNA by methionine aminoacyl tRNA synthetase. Nucleic Acids Res. 2021, 49, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Nallar, S.C.; Lin, L.; Srivastava, V.; Gade, P.; Hofmann, E.R.; Ahmed, H.; Reddy, S.P.; Kalvakolanu, D.V. GRIM-1, a novel growth suppressor, inhibits rRNA maturation by suppressing small nucleolar RNAs. PLoS ONE 2011, 6, e24082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieron, P.; Hader, C.; Hatina, J.; Engers, R.; Wlazlinski, A.; Muller, M.; Schulz, W.A. DKC1 overexpression associated with prostate cancer progression. Br. J. Cancer 2009, 101, 1410–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katunaric, M.; Zamolo, G. Modulating telomerase activity in tumor patients by targeting dyskerin binding site for hTR. Med. Hypotheses 2012, 79, 319–320. [Google Scholar] [CrossRef] [PubMed]
- Stockert, J.A.; Gupta, A.; Herzog, B.; Yadav, S.S.; Tewari, A.K.; Yadav, K.K. Predictive value of pseudouridine in prostate cancer. Am. J. Clin. Exp. Urol. 2019, 7, 262–272. [Google Scholar]
- McMahon, M.; Contreras, A.; Ruggero, D. Small RNAs with big implications: New insights into H/ACA snoRNA function and their role in human disease. Wiley Interdiscip. Rev. RNA 2015, 6, 173–189. [Google Scholar] [CrossRef] [Green Version]
- Martens-Uzunova, E.S.; Jalava, S.E.; Dits, N.F.; van Leenders, G.J.; Moller, S.; Trapman, J.; Bangma, C.H.; Litman, T.; Visakorpi, T.; Jenster, G. Diagnostic and prognostic signatures from the small non-coding RNA transcriptome in prostate cancer. Oncogene 2012, 31, 978–991. [Google Scholar] [CrossRef] [Green Version]
- Crea, F.; Quagliata, L.; Michael, A.; Liu, H.H.; Frumento, P.; Azad, A.A.; Xue, H.; Pikor, L.; Watahiki, A.; Morant, R.; et al. Integrated analysis of the prostate cancer small-nucleolar transcriptome reveals SNORA55 as a driver of prostate cancer progression. Mol. Oncol. 2016, 10, 693–703. [Google Scholar] [CrossRef]
- Gong, J.; Li, Y.; Liu, C.J.; Xiang, Y.; Li, C.; Ye, Y.; Zhang, Z.; Hawke, D.H.; Park, P.K.; Diao, L.; et al. A Pan-cancer Analysis of the Expression and Clinical Relevance of Small Nucleolar RNAs in Human Cancer. Cell Rep. 2017, 21, 1968–1981. [Google Scholar] [CrossRef] [Green Version]
- Jana, S.; Hsieh, A.C.; Gupta, R. Reciprocal amplification of caspase-3 activity by nuclear export of a putative human RNA-modifying protein, PUS10 during TRAIL-induced apoptosis. Cell Death Dis. 2017, 8, e3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, A.; Huang, V.; Livingstone, J.; Wang, J.; Fox, N.S.; Kurganovs, N.; Ignatchenko, V.; Fritsch, K.; Donmez, N.; Heisler, L.E.; et al. The Proteogenomic Landscape of Curable Prostate Cancer. Cancer Cell 2019, 35, 414–427.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Rambla, C.; Puchades-Carrasco, L.; García-Flores, M.; Rubio-Briones, J.; López-Guerrero, J.A.; Pineda-Lucena, A. Non-invasive urinary metabolomic profiling discriminates prostate cancer from benign prostatic hyperplasia. Metabolomics 2017, 13, 52. [Google Scholar] [CrossRef] [PubMed]
- Christofi, T.; Zaravinos, A. RNA editing in the forefront of epitranscriptomics and human health. J. Transl. Med. 2019, 17, 319. [Google Scholar] [CrossRef]
- Mo, F.; Wyatt, A.W.; Sun, Y.; Brahmbhatt, S.; McConeghy, B.J.; Wu, C.; Wang, Y.; Gleave, M.E.; Volik, S.V.; Collins, C.C. Systematic identification and characterization of RNA editing in prostate tumors. PLoS ONE 2014, 9, e101431. [Google Scholar] [CrossRef] [Green Version]
- Paz-Yaacov, N.; Bazak, L.; Buchumenski, I.; Porath, H.T.; Danan-Gotthold, M.; Knisbacher, B.A.; Eisenberg, E.; Levanon, E.Y. Elevated RNA Editing Activity Is a Major Contributor to Transcriptomic Diversity in Tumors. Cell Rep. 2015, 13, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Beyer, U.; Brand, F.; Martens, H.; Weder, J.; Christians, A.; Elyan, N.; Hentschel, B.; Westphal, M.; Schackert, G.; Pietsch, T.; et al. Rare ADAR and RNASEH2B variants and a type I interferon signature in glioma and prostate carcinoma risk and tumorigenesis. Acta Neuropathol. 2017, 134, 905–922. [Google Scholar] [CrossRef] [Green Version]
- Martinez, H.D.; Jasavala, R.J.; Hinkson, I.; Fitzgerald, L.D.; Trimmer, J.S.; Kung, H.J.; Wright, M.E. RNA editing of androgen receptor gene transcripts in prostate cancer cells. J. Biol. Chem. 2008, 283, 29938–29949. [Google Scholar] [CrossRef] [Green Version]
- Barros-Silva, D.; Klavert, J.; Jenster, G.; Jeronimo, C.; Lafontaine, D.L.J.; Martens-Uzunova, E.S. The role of OncoSnoRNAs and Ribosomal RNA 2’-O-methylation in Cancer. RNA Biol. 2021, 18, 61–74. [Google Scholar] [CrossRef]
- Pacilli, A.; Ceccarelli, C.; Trere, D.; Montanaro, L. SnoRNA U50 levels are regulated by cell proliferation and rRNA transcription. Int. J. Mol. Sci. 2013, 14, 14923–14935. [Google Scholar] [CrossRef] [Green Version]
- Yi, Y.; Li, Y.; Meng, Q.; Li, Q.; Li, F.; Lu, B.; Shen, J.; Fazli, L.; Zhao, D.; Li, C.; et al. A PRC2-independent function for EZH2 in regulating rRNA 2’-O methylation and IRES-dependent translation. Nat. Cell Biol. 2021, 23, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Seruga, B.; Ocana, A.; Tannock, I.F. Drug resistance in metastatic castration-resistant prostate cancer. Nat. Rev. Clin. Oncol. 2011, 8, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Munster, P.N.; Marchion, D.; Thomas, S.; Egorin, M.; Minton, S.; Springett, G.; Lee, J.H.; Simon, G.; Chiappori, A.; Sullivan, D.; et al. Phase I trial of vorinostat and doxorubicin in solid tumours: Histone deacetylase 2 expression as a predictive marker. Br. J. Cancer 2009, 101, 1044–1050. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Meng, S.; Xu, M.; Wang, S.; He, L.; Xu, X.; Wang, X.; Xie, L. Downregulation of N(6)-methyladenosine binding YTHDF2 protein mediated by miR-493-3p suppresses prostate cancer by elevating N(6)-methyladenosine levels. Oncotarget 2017, 9, 3752–3764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladang, A.; Rapino, F.; Heukamp, L.C.; Tharun, L.; Shostak, K.; Hermand, D.; Delaunay, S.; Klevernic, I.; Jiang, Z.; Jacques, N.; et al. Elp3 drives Wnt-dependent tumor initiation and regeneration in the intestine. J. Exp. Med. 2015, 212, 2057–2075. [Google Scholar] [CrossRef] [Green Version]



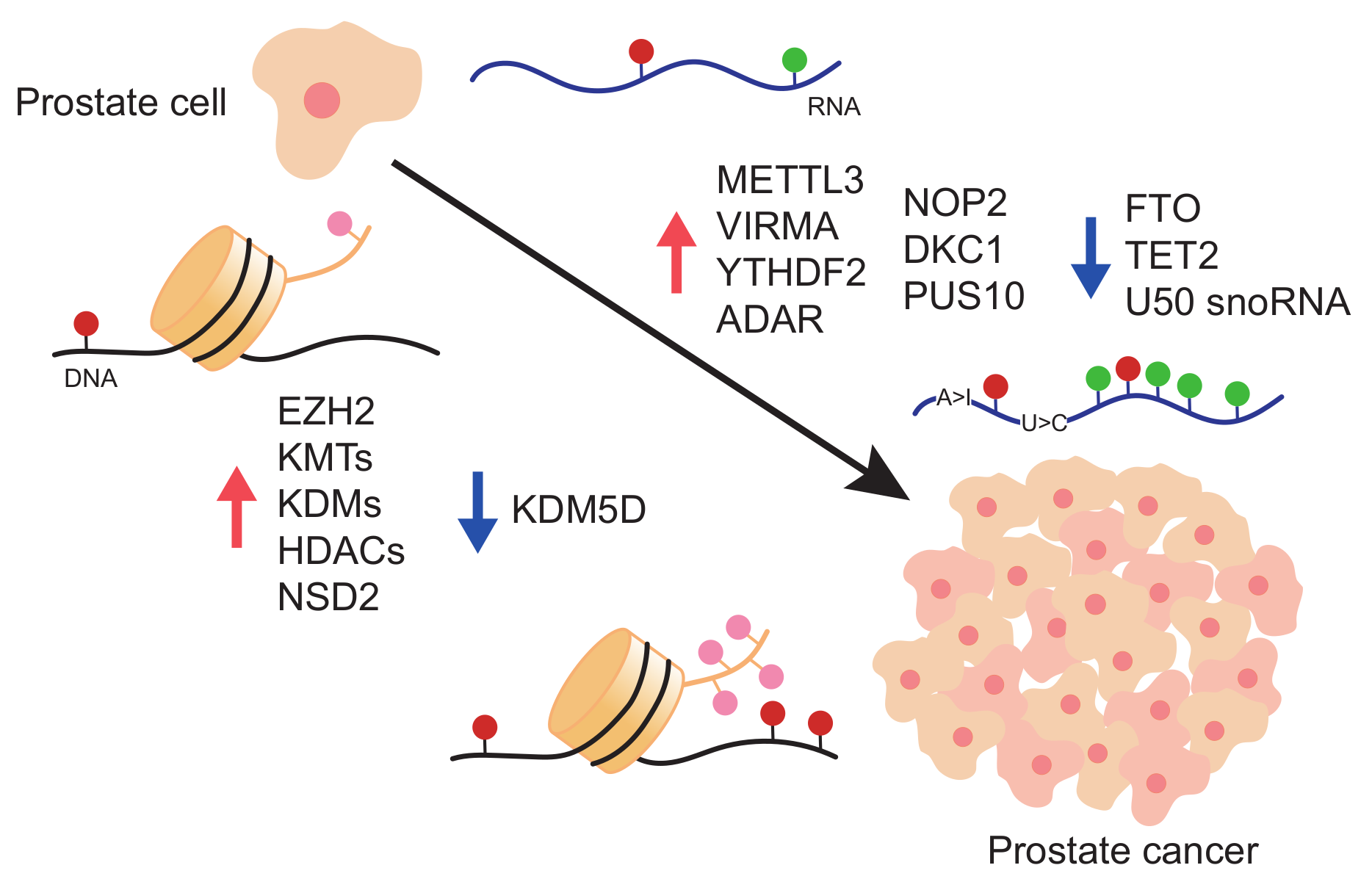{kind=link}
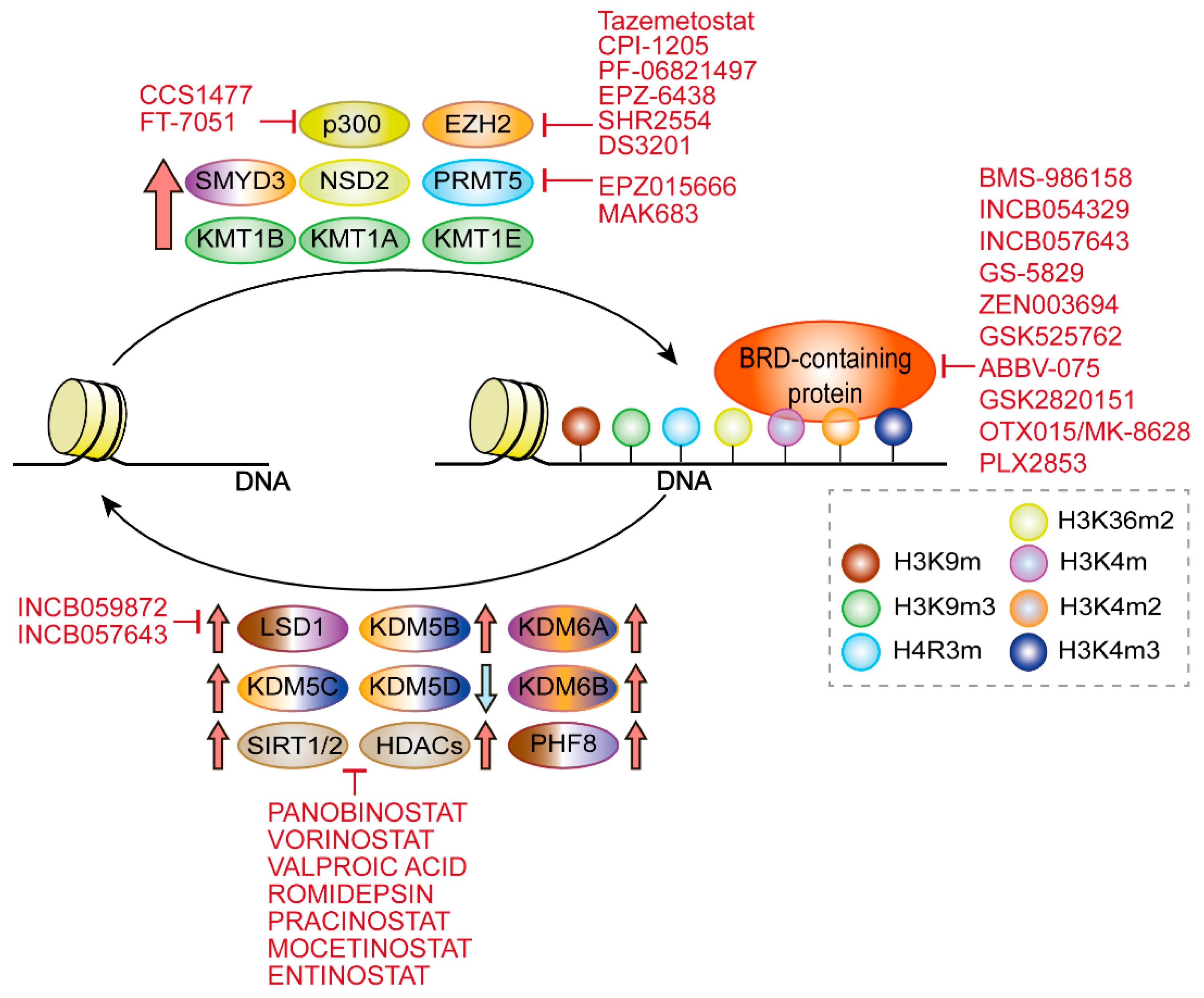{kind=link}
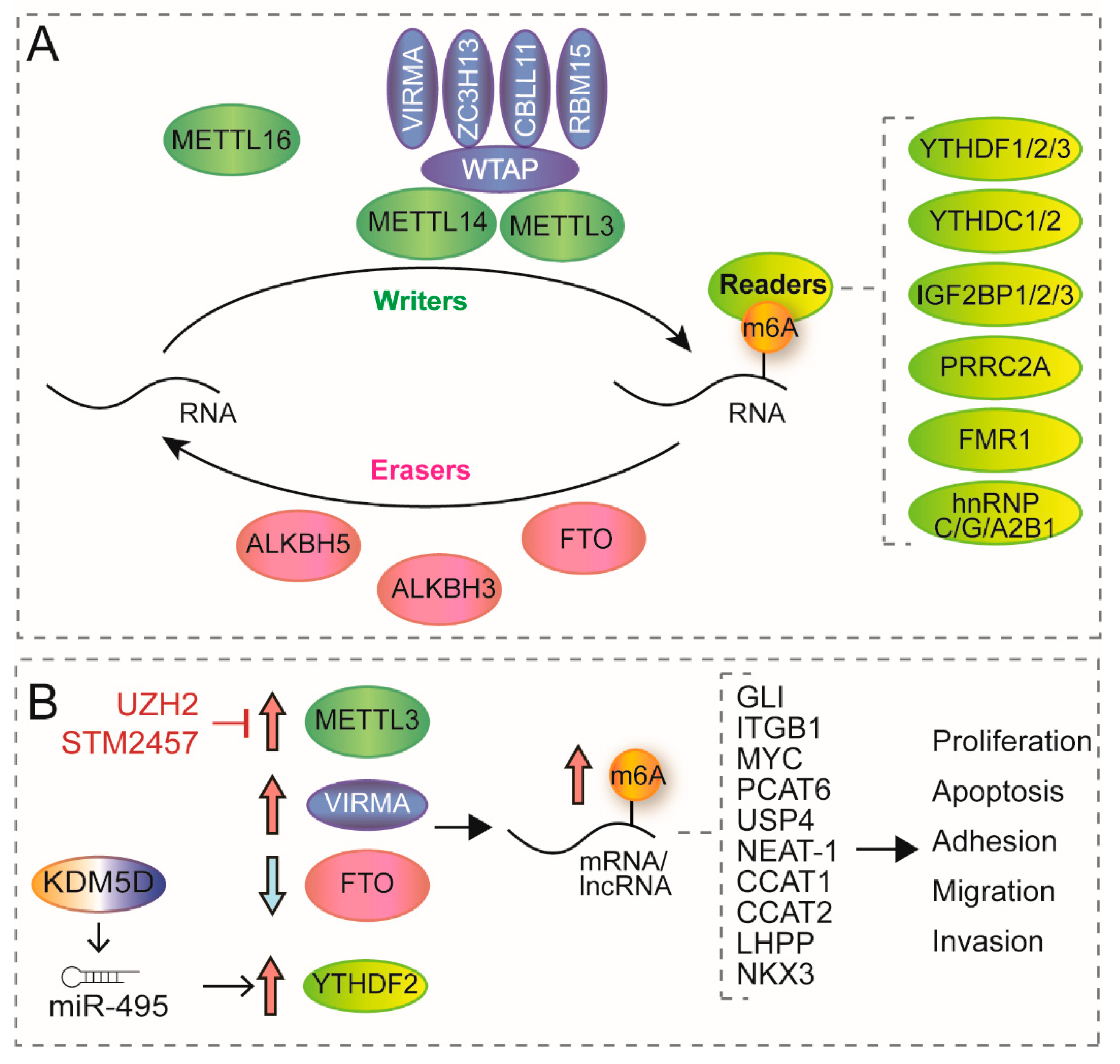{kind=link}
| DRUG | TRAIL ID | PHASE | PROTOCOL | STATUS |
|---|---|---|---|---|
| DNMT INHIBITORS | ||||
| 5-AZACYTIDINE | NCT00384839 | Phase II | Patients with CRPC received 75 mg/m2 of 5-azacytidine for five consecutive days of a 28-day cycle. Patients were treated until clinical progression up to a maximum of 12 cycles. | Completed. 5-Azacytidine modulates PSA (doubling time > 3 months) in 56% of patients. Clinical progression-free survival of 12.4 weeks [14] |
| 5-AZACYTIDINE | NCT00503984 | Phase I/II | mCRPC (+docetaxel, prednisone) | Completed [15]. |
| 5-AZACYTIDINE | NCT00006019 | Phase II | mCRPC (+ Sodium phenylbutyrate) | Completed. |
| DISULFIRAM | NCT01118741 | CRPC | Completed. | |
| DECITABINE | NCT03709550 | Phase I/II | mCRPC (+Enzalutamide) | Not yet recruiting. |
| 5-AZACYTIDINE | NCT02959437 | Phase I/II | Advanced Solid tumours (+ PD-1 + IDO-1) | Terminated by Sponsor |
| HMT INHIBITORS | ||||
| PRMT5 INHIBITOR MAK683 | NCT02900651 | Phase I/II | Diffuse large B cell lymphoma, advanced solid tumours | Recruiting |
| EZH2 INHIBITOR TAZEMETOSTAT | NCT03213665 | Phase II | Advanced solid tumours | Active. Not Recruiting |
| EZH2 INHIBITOR CPI-1205 | NCT03480646 | Phase I/II | mCRPC (+Abiraterone/prednisone or enzalutamide) | Active. Not Recruiting |
| EZH2 INHIBITOR PF-06821497 | NCT03460977 | Phase I | mCRPC | Recruiting |
| EZH2 INHIBITOR EPZ-6438 | NCT04179864 | Phase Ib | mCRPC (+Abiraterone/prednisone or enzalutamide) | Recruiting |
| EZH2 INHIBITOR SHR2554 | NCT03741712 | Phase I/II | mCRPC (+SHR3680) | Terminated |
| EZH1/2 INHIBITOR DS3201 | NCT04388852 | Phase I/II | mCRPC (+Ipilimumab) | Recruiting |
| HAT INHIBITORS | ||||
| P300/CBP INHIBITOR CCS1477 | NCT03568656 | Phase I/II | mCRPC (+Abiraterone/prednisone or enzalutamide) | Recruiting |
| P300/CBP INHIBITOR: FT-7051 | NCT04575766 | Phase I | mCRPC | Recruiting |
| BRD-CONTAINING PROTEIN INHIBITORS | ||||
| BMS-986158 | NCT02419417 | Phase I/II | Advanced solid tumours | Active. Not Recruiting |
| INCB054329 | NCT02431260 | Phase I/II | Advanced solid tumours | Terminated |
| INCB057643 | NCT02711137 | Phase I/II | Advanced solid tumours (+abiraterone) | Terminated |
| GS-5829 | NCT02607228 | Phase I/II | mCRPC (+enzalutamide) | Terminated |
| ZEN003694 | NCT02711956 | Phase I/II | mCRPC (+enzalutamide) | Completed. Longer PFS in a subset of patients [16]. |
| ZEN003694 | NCT02705469 | Phase I | mCRPC | Completed. |
| ZEN003694 | NCT04471974 | Phase II | mCRPC (+Enzalutamide + pembrolizumab) | Recruiting |
| GSK525762 | NCT03150056 | Phase I | mCRPC (+Abiraterone/prednisone or enzalutamide) | Completed |
| ABBV-075 | NCT02391480 | Phase I | mCRPC | Completed. Not significant antitumour activity [17]. |
| GSK2820151 | NCT02630251 | Phase I | Advanced or recurrent solid tumours | Terminated (In 2017, GSK2820151 was terminated due to development of another BET Inhibitor (GSK525762) with a better understanding of the risk benefit profile.) |
| OTX015/MK-8628 | NCT02259114 | Phase Ib | mCPRC | Completed. Not significant antitumour activity [18]. |
| PLX2853 | NCT04556617 | Phase I/II | mCPRC (+Abiraterone/prednisone or olaparib) | Recruiting |
| HDMT INHIBITORS | ||||
| LSD1 INHIBITOR: INCB059872 | NCT02712905 | Phase I/II | Solid tumours and hematologic malignancy | Active. Not Recruiting |
| LSD1 INHIBITOR: INCB059872 | NCT02959437 | Phase I/II | Advanced Solid tumours (+pembrolizumab + epacadostat) | Terminated by Sponsor |
| LSD1 INHIBITOR: INCB057643 | NCT02959437 | Phase I/II | Advanced Solid tumours (+pembrolizumab + epacadostat) | Terminated by Sponsor |
| HDAC INHIBITORS | ||||
| VORINOSTAT/SAHA | NCT00005634 | Phase I | mCRPC | Completed. Determine the tolerability, pharmacokinetic profile, and biological effects of the drug. Not available [19]. |
| VORINOSTAT/SAHA | NCT00330161 | Phase II | mCRPC with disease progression on prior chemotherapy received 400 mg vorinostat/SAHA orally each day. Disease progression measured at 6 months. n = 27 | Completed. Toxicity: significant toxicities including fatigue, nausea. IL-6 was higher in patients with toxicity. Seven percent of patients achieved a stable disease state. No PSA decline >50% observed. Median time to progression and overall survival were 2.8 and 11.7 months, respectively. Significant toxicities reported [19]. |
| VORINOSTAT/SAHA AND DOCETAXEL | NCT00565227 | Phase I | Patients with advanced and relapsed tumours received oral vorinostat/SAHA for the first 14 days of a 21-day cycle, with docetaxel I.v. on day 4 of each cycle. n = 12 | Completed. Toxicity: neutropenia, peripheral neuropathy, and gastrointestinal bleeding. The combination of vorinostat/SAHA and docetaxel was poorly tolerated. No responses were identified [20]. |
| VORINOSTAT/SAHA | NCT00589472 | Phase II | Localised PCa (+Bicalutamide, goserelin acetate, or leuprolide acetate) | Completed. |
| VALPROIC ACID | NCT00530907 | Phase I | CRPC (+Bevacizumab) | Completed [21]. |
| PANOBINOSTAT (LBH589) | NCT00667862 | Phase II | I.v. panobinostat (20 mg/m2) was administered to CRPC patients on days 1 and 8 of a 21-day cycle. Disease progression measured at 24 weeks. n = 35 | Completed. Toxicity: fatigue, thrombocytopenia, nausea; 14% of patients demonstrated a decrease in PSA but none >50%. No clinical activity [22]. |
| PANOBINOSTAT | NCT00878436 | Phase I/II | CRPC (+bicalutamide) | Completed [23]. |
| PANOBINOSTAT, DOCETAXEL, AND PREDNISONE | NCT00663832 | Phase I | CRPC patients received oral panobinostat (20 mg/m2) the first, third and fifth day of the week for 2 consecutive weeks. In addition, patients received oral Panobinostat (15 mg/m2) with docetaxel I.v. (75 mg/m2) every 21 days and oral prednisone (5 mg) twice every day of a 21-day cycle. n = 16 | Completed. Toxicity: dyspnoea and neutropenia The combination in patients with CRPC resulted in 63% of patients with >50% decline in PSA levels. No relevant anti-tumour activity [24]. |
| PANOBINOSTAT DOCETAXEL AND PREDNISONE | NCT00493766 | Phase I | On the one hand, oral panobinostat alone is given to patients with progressing hormone refractory prostate cancer. On the other hand, oral Panobinostat along with I.v. docetaxel and oral prednisone is administered. n = 16 | Completed. Toxicity: dyspnoea, neutropenia, fatigue. Exposure to oral panobinostat was similar with and without docetaxel [25]. |
| PANOBINOSTAT | NCT00670553 | Phase I | Localised prostate cancer (+External beam radiotherapy) | Completed. |
| PANOBINOSTAT | NCT00667862 | Phase I | mCRPC | Completed. |
| ROMIDEPSIN | NCT00106418 | Phase II | mCRPC patients received romidepsin (13 mg/m2) intravenously on days 1, 8, and 15 every 21-day cycle. Disease progression measures at 6 months. n = 35 | Completed. Toxicity: nausea, fatigue. Two patients reached a confirmed radiological partial response of over 6 months, in addition to >50% PSA decline. Eleven patients had to discontinue the therapy due to toxicity. Romidepsin demonstrated minimal anti-tumour activity in chemonaive patients with CRPC [26] |
| PRACINOSTAT (SB939) | NCT01075308 | Phase II | mCRPC | Completed [27]. |
| MOCETINOSTAT (MGCD0103) | NCT00511576 | Phase I | Advanced cancer tumours | Celgene terminated its collaboration agreement with MethylGene for the development of MGCD0103. |
| ENTINOSTAT (MS-275) | NCT03829930 | Phase I | CRPC (+Enzalutamide) | Terminated (Sponsor discontinued the drug). |
| ENTINOSTAT (MS-275) | NCT00020579 | Phase I | Advanced solid tumours or lymphoma. | Completed [28]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López, J.; Añazco-Guenkova, A.M.; Monteagudo-García, Ó.; Blanco, S. Epigenetic and Epitranscriptomic Control in Prostate Cancer. Genes 2022, 13, 378. https://doi.org/10.3390/genes13020378
López J, Añazco-Guenkova AM, Monteagudo-García Ó, Blanco S. Epigenetic and Epitranscriptomic Control in Prostate Cancer. Genes. 2022; 13(2):378. https://doi.org/10.3390/genes13020378
Chicago/Turabian StyleLópez, Judith, Ana M. Añazco-Guenkova, Óscar Monteagudo-García, and Sandra Blanco. 2022. "Epigenetic and Epitranscriptomic Control in Prostate Cancer" Genes 13, no. 2: 378. https://doi.org/10.3390/genes13020378