
A Split NanoLuc Reporter Quantitatively Measures Circular RNA IRES Translation

 ,
,  and
and 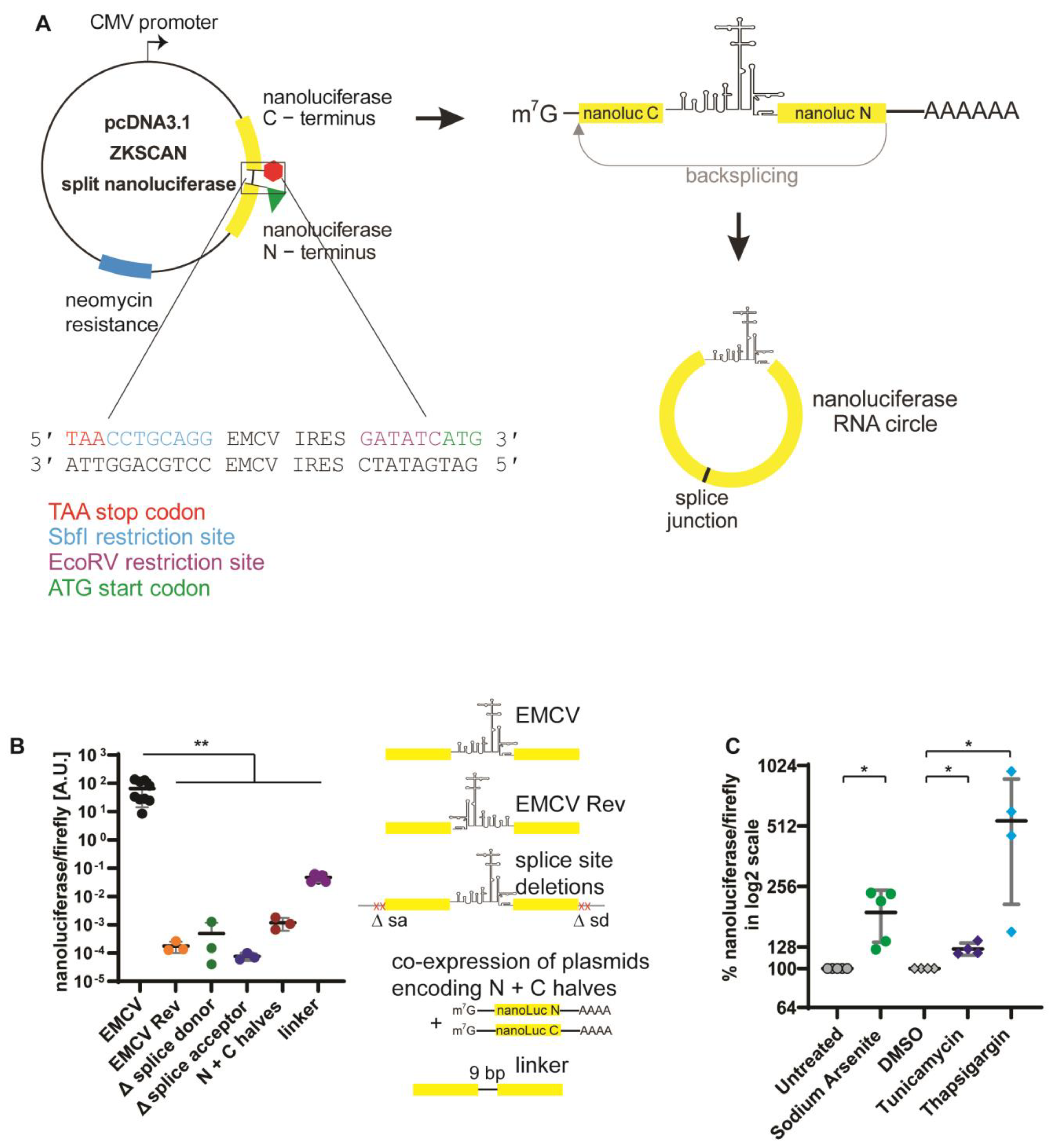{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning
2.2. Cell Culture
2.3. Luciferase Assay
3. Results
3.1. Development of a Split NanoLuc Reporter
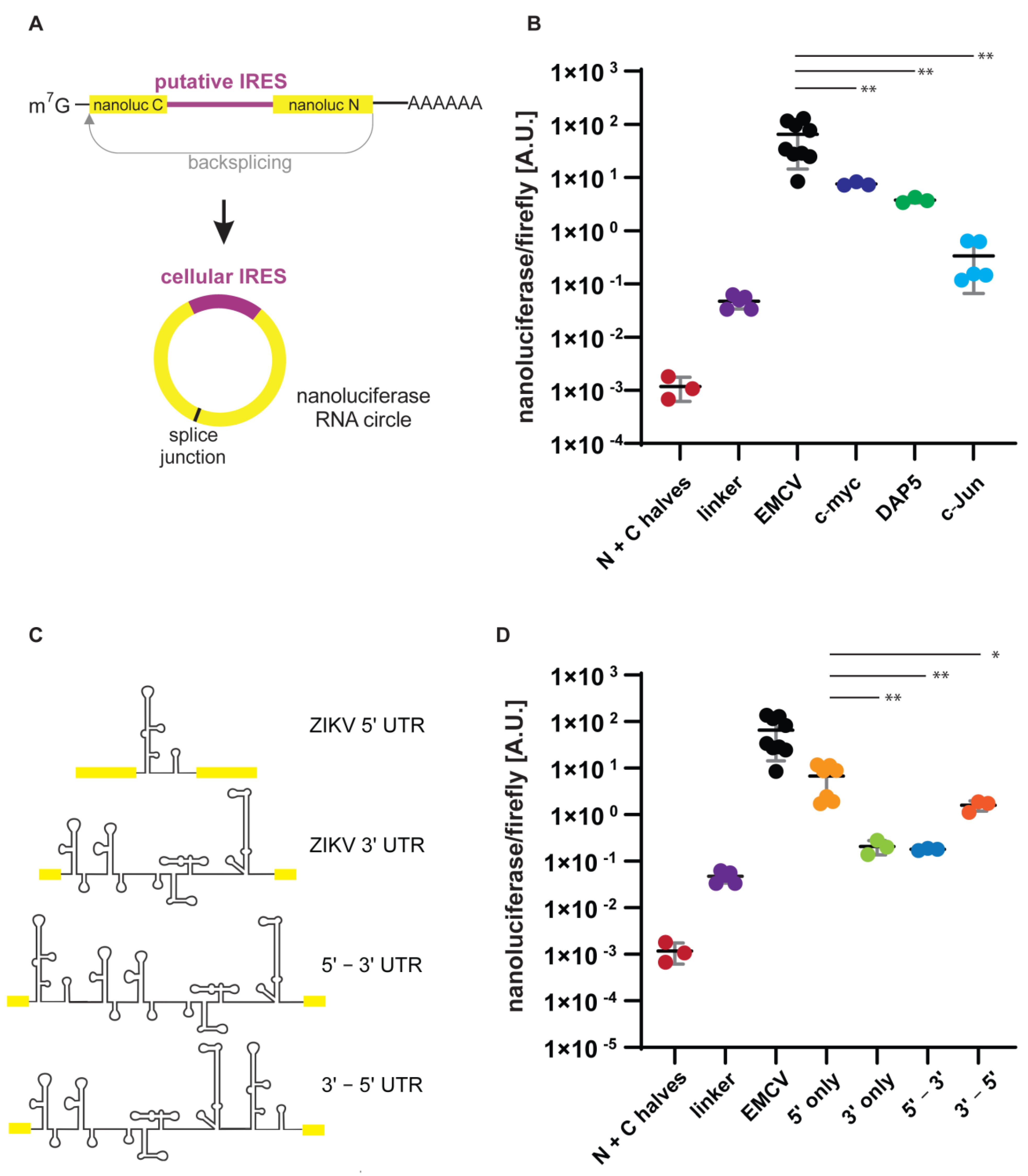
3.2. Putative IRESs
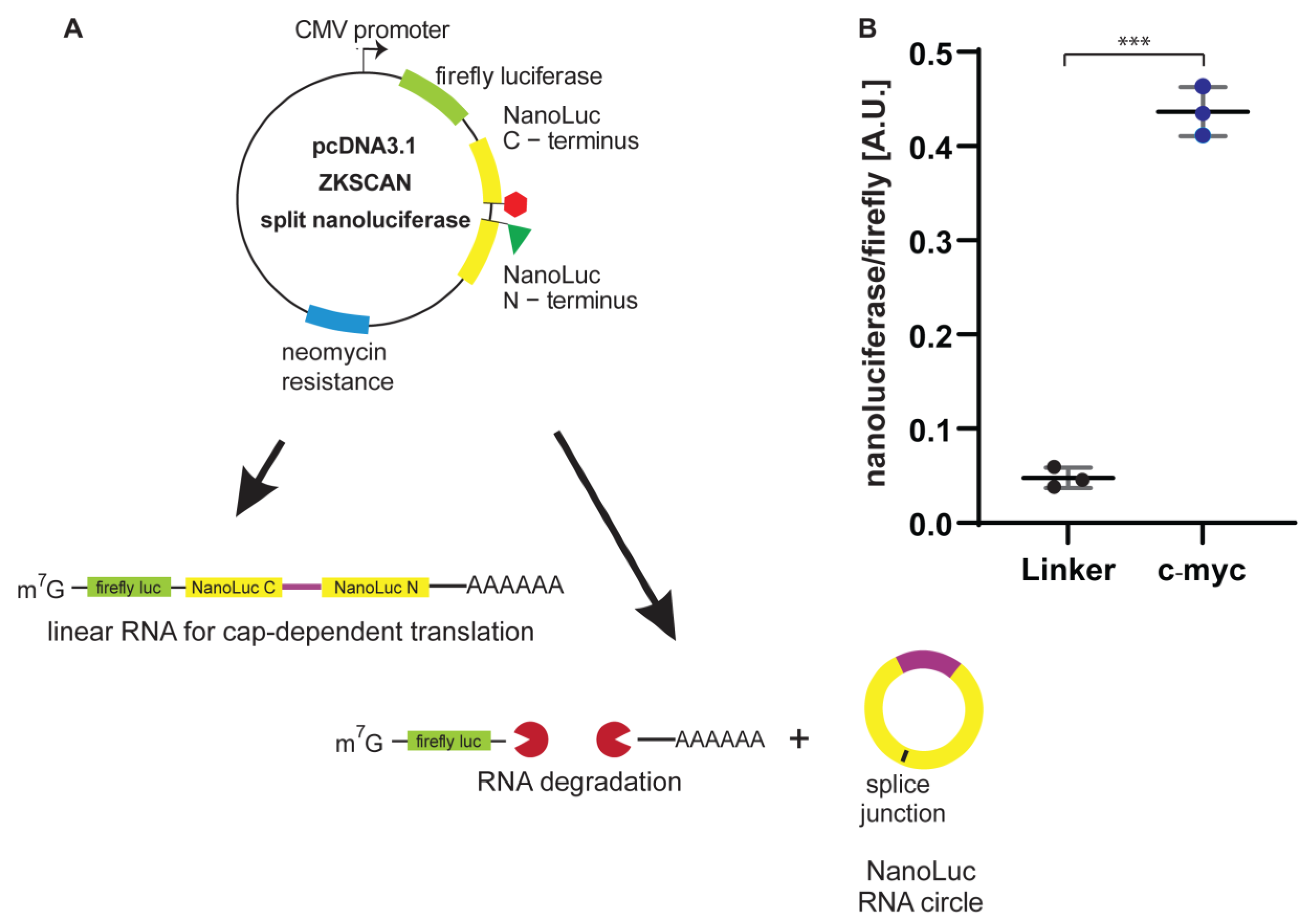
3.3. Generation of a Single-Plasmid, Dual-Luciferase Reporter
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ramanathan, A.; Robb, G.B.; Chan, S.-H. mRNA capping: Biological functions and applications. Nucleic Acids Res. 2016, 44, 7511–7526. [Google Scholar] [CrossRef] [PubMed]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merrick, W.C.; Pavitt, G.D. Protein Synthesis Initiation in Eukaryotic Cells. Cold Spring Harb. Perspect. Biol. 2018, 10, a033092. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.; Graff, J.; Ruggero, D.; Sonenberg, N. Targeting the eIF4F translation initiation complex: A critical nexus for cancer development. Cancer Res. 2015, 75, 250–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robichaud, N.; Sonenberg, N.; Ruggero, D.; Schneider, R.J. Translational Control in Cancer. Cold Spring Harb. Perspect. Biol. 2018, 11, a032896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.-M.; Chen, C.-J.; Shih, S.-R. Regulation Mechanisms of Viral IRES-Driven Translation. Trends Microbiol. 2017, 25, 546–561. [Google Scholar] [CrossRef] [PubMed]
- Mailliot, J.; Martin, F. Viral internal ribosomal entry sites: Four classes for one goal. Wiley Interdiscip. Rev. RNA 2018, 9, e1458. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.K.; Krausslich, H.G.; Nicklin, M.J.H.; Duke, G.M.; Palmenberg, A.C.; Wimmer, E. A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J. Virol. 1988, 62, 2636–2643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrenfeld, E. Initiation of translation by picornavirus RNAs. In Translational Control; Hershey, J.W.B., Mathews, M.B., Sonenberg, N., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1996; pp. 549–573. [Google Scholar]
- Balvay, L.; Rifo, R.S.; Ricci, E.P.; Decimo, D.; Ohlmann, T. Structural and functional diversity of viral IRESes. Biochim. Biophys. Acta BBA-Gene Regul. Mech. 2009, 1789, 542–557. [Google Scholar] [CrossRef] [PubMed]
- Kuyumcu-Martinez, N.M.; Joachims, M.; Lloyd, R.E. Efficient cleavage of ribosome-associated poly(A)-binding protein by enterovirus 3C protease. J. Virol. 2002, 76, 2062–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuyumcu-Martinez, N.M.; van Eden, M.E.; Younan, P.; Lloyd, R.E. Cleavage of poly(A)-binding protein by poliovirus 3C protease inhibits host cell translation: A novel mechanism for host translation shutoff. Mol. Cell. Biol. 2004, 24, 1779–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventoso, I.; MacMillan, S.E.; Hershey, J.W.; Carrasco, L. Poliovirus 2A proteinase cleaves directly the eIF-4G subunit of eIF-4F complex. FEBS Lett. 1998, 435, 79–83. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Salas, E. The impact of RNA structure on picornavirus IRES activity. Trends Microbiol. 2008, 16, 230–237. [Google Scholar] [CrossRef]
- Johannes, G.; Carter, M.S.; Eisen, M.B.; Brown, P.O.; Sarnow, P. Identification of eukaryotic mRNAs that are translated at reduced cap binding complex eIF4F concentrations using a cDNA microarray. Proc. Natl. Acad. Sci. USA 1999, 96, 13118–13123. [Google Scholar] [CrossRef] [Green Version]
- Carter, M.S.; Kuhn, K.M.; Sarnow, P. Cellular internal ribosome entry site elements and the use of cDNA microarray in their investigation. In Translational Control of Gene Expression; Sonenberg, N., Hershey, J.W.B., Mathwes, M.B., Eds.; Cold Spring Harbor Press: Cold Spring Harbor, NY, USA, 2000; pp. 615–635. [Google Scholar]
- Jackson, R.J. The current status of vertebrate cellular mRNA IRESs. Cold Spring Harb. Perspect. Biol. 2013, 5, a011569. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Komar, A.A.; Hatzoglou, M. Cellular IRES-Mediated Translation: The War of ITAFs in Pathophysiological States; Taylor and Francis Inc.: Philadelphia, PA, USA, 2011; Volume 10. [Google Scholar]
- Lewis, S.M.; Cerquozzi, S.; Graber, T.E.; Ungureanu, N.H.; Andrews, M.; Holcik, M. The eIF4G homolog DAP5/p97 supports the translation of select mRNAs during endoplasmic reticulum stress. Nucleic Acids Res. 2008, 36, 168–178. [Google Scholar] [CrossRef] [Green Version]
- Thompson, S.R. So You Want to Know If Your Message Has an IRES? Wiley Interdiscip Rev RNA: Wiley Hoboken, NJ, USA, 2012; Volume 3. [Google Scholar]
- Chen, C.Y.; Sarnow, P. Internal ribosome entry sites tests with circular mRNAs. Methods Mol. Biol. 1998, 77, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Appel, B. In vitro circularization of RNA. RNA Biol. 2017, 14, 1018–1027. [Google Scholar] [CrossRef]
- Lasda, E.; Parker, R. Circular RNAs: Diversity of form and function. RNA 2014, 20, 1829–1842. [Google Scholar] [CrossRef] [Green Version]
- Wilusz, J.E. Circular RNAs: Unexpected outputs of many protein-coding genes. RNA Biol. 2017, 14, 1007–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.-L. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat. Rev. Mol. Cell Biol. 2020, 21, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.C.; Liang, D.; Tatomer, D.C.; Gold, B.; March, Z.M.; Cherry, S.; Wilusz, J.E. Combinatorial control of Drosophila circular RNA expression by intronic repeats, hnRNPs, and SR proteins. Genes Dev. 2015, 29, 2168–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, D.; Wilusz, J.E. Short intronic repeat sequences facilitate circular RNA production. Genes Dev. 2014, 28, 2233–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNabb, D.S.; Reed, R.; Marciniak, R.A. Dual luciferase assay system for rapid assessment of gene expression in Saccharomyces cerevisiae. Eukaryot. Cell 2005, 4, 1539–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- England, C.G.; Ehlerding, E.B.; Cai, W. NanoLuc: A Small Luciferase Is Brightening Up the Field of Bioluminescence. Bioconjug. Chem. 2016, 27, 1175–1187. [Google Scholar] [CrossRef]
- Zhao, J.; Nelson, T.J.; Vu, Q.; Truong, T.; Stains, C.I. Self-Assembling NanoLuc Luciferase Fragments as Probes for Protein Aggregation in Living Cells. ACS Chem. Biol. 2016, 11, 132–138. [Google Scholar] [CrossRef] [Green Version]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [Green Version]
- Wek, R.C. Role of eIF2α Kinases in Translational Control and Adaptation to Cellular Stress. Cold Spring Harb. Perspect. Biol. 2018, 10, a032870. [Google Scholar] [CrossRef]
- Stoneley, M.; Paulin, F.E.; Le Quesne, J.P.; Chappell, S.A.; Willis, A.E. C-Myc 5′ untranslated region contains an internal ribosome entry segment. Oncogene 1998, 16, 423–428. [Google Scholar] [CrossRef] [Green Version]
- Johannes, G.; Sarnow, P. Cap-independent polysomal association of natural mRNAs encoding c-myc, BiP, and eIF4G conferred by internal ribosome entry sites. Rna 1998, 4, 1500–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehgal, A.; Briggs, J.; Rinehart-Kim, J.; Basso, J.; Bos, T.J. The chicken c-Jun 5′ untranslated region directs translation by internal initiation. Oncogene 2000, 19, 2836–2845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blau, L.; Knirsh, R.; Ben-Dror, I.; Oren, S.; Kuphal, S.; Hau, P.; Proescholdt, M.; Bosserhoff, A.-K.; Vardimon, L. Aberrant expression of c-Jun in glioblastoma by internal ribosome entry site (IRES)-mediated translational activation. Proc. Natl. Acad. Sci. USA 2012, 109, E2875–E2884. [Google Scholar] [CrossRef] [Green Version]
- Edgil, D.; Polacek, C.; Harris, E. Dengue virus utilizes a novel strategy for translation initiation when cap-dependent translation is inhibited. J. Virol. 2006, 80, 2976–2986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Mugavero, J.A.; Stauft, C.B.; Wimmer, E. Dengue and zika virus 5′ untranslated regions harbor internal ribosomal entry site functions. MBio 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Fan, X.; Mao, M.; Song, X.; Wu, P.; Zhang, Y.; Jin, Y.; Yang, Y.; Chen, L.-L.; Wang, Y.; et al. Extensive translation of circular RNAs driven by N6-methyladenosine. Cell Res. 2017, 27, 626–641. [Google Scholar] [CrossRef] [Green Version]
- Weingarten-Gabbay, S.; Elias-Kirma, S.; Nir, R.; Gritsenko, A.A.; Stern-Ginossar, N.; Yakhini, Z.; Weinberger, A.; Segal, E. Comparative genetics. Systematic discovery of cap-independent translation sequences in human and viral genomes. Science 2016, 351, aad4939. [Google Scholar] [CrossRef]
- Matsuda, D.; Mauro, V.P. Base pairing between hepatitis C virus RNA and 18S rRNA is required for IRES-dependent translation initiation in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 15385–15389. [Google Scholar] [CrossRef] [Green Version]
- Thoma, C.; Bergamini, G.; Galy, B.; Hundsdoerfer, P.; Hentze, M.W. Enhancement of IRES-mediated translation of the c-myc and BiP mRNAs by the poly(A) tail is independent of intact eIF4G and PABP. Mol. Cell 2004, 15, 925–935. [Google Scholar] [CrossRef]
- Henis-Korenblit, S.; Strumpf, N.L.; Goldstaub, D.; Kimchi, A. A novel form of DAP5 protein accumulates in apoptotic cells as a result of caspase cleavage and internal ribosome entry site-mediated translation. Mol. Cell. Biol. 2000, 20, 496–506. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.S.Y.; Kranzusch, P.J.; Doudna, J.A.; Cate, J.H.D. eIF3d is an mRNA cap-binding protein that is required for specialized translation initiation. Nature 2016, 536, 96–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, M.J.; Shortridge, M.D.; Albin, D.D.; Cominsky, L.Y.; Varani, G. Structure of the RNA Specialized Translation Initiation Element that Recruits eIF3 to the 5′-UTR of c-Jun. J. Mol. Biol. 2020, 432, 1841–1855. [Google Scholar] [CrossRef] [PubMed]
- Meganck, R.M.; Liu, J.; Hale, A.E.; Simon, K.E.; Fanous, M.M.; Vincent, H.A.; Wilusz, J.E.; Moorman, N.J.; Marzluff, W.F.; Asokan, A. Engineering highly efficient backsplicing and translation of synthetic circRNAs. Mol. Ther. Nucleic Acids 2021, 23, 821–834. [Google Scholar] [CrossRef]
- Fernandez, J.; Yaman, I.; Sarnow, P.; Snider, M.D.; Hatzoglou, M. Regulation of internal ribosomal entry site-mediated translation by phosphorylation of the translation initiation factor eIF2alpha. J. Biol. Chem. 2002, 277, 19198–19205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marceau, C.D.; Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Brewer, S.M.; Fuchs, G.; Swaminathan, K.; Mata, M.A.; Elias, J.E.; Sarnow, P.; et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 2016, 535, 159–163. [Google Scholar] [CrossRef] [Green Version]
- Hafirassou, M.L.; Meertens, L.; Umaña-Diaz, C.; Labeau, A.; Dejarnac, O.; Bonnet-Madin, L.; Kümmerer, B.M.; Delaugerre, C.; Roingeard, P.; Vidalain, P.-O.; et al. A Global Interactome Map of the Dengue Virus NS1 Identifies Virus Restriction and Dependency Host Factors. Cell Rep. 2017, 21, 3900–3913. [Google Scholar] [CrossRef] [Green Version]
- Petrova, E.; Gracias, S.; Beauclair, G.; Tangy, F.; Jouvenet, N. Uncovering flavivirus host dependency factors through a genome-wide gain-of-function screen. Viruses 2019, 11, 68. [Google Scholar] [CrossRef] [Green Version]
- Landry, D.M.; Hertz, M.I.; Thompson, S.R. RPS25 is essential for translation initiation by the Dicistroviridae and hepatitis C viral IRESs. Genes Dev. 2009, 23, 2753–2764. [Google Scholar] [CrossRef] [Green Version]
- Hertz, M.I.; Landry, D.M.; Willis, A.E.; Luo, G.; Thompson, S.R. Ribosomal protein s25 dependency reveals a common mechanism for diverse internal ribosome entry sites and ribosome shunting. Mol. Cell. Biol. 2013, 33, 1016–1026. [Google Scholar] [CrossRef] [Green Version]
- Fernández-García, L.; Angulo, J.; Ramos, H.; Barrera, A.; Pino, K.; Vera-Otarola, J.; López-Lastra, M. The internal ribosome entry site of the Dengue virus mRNA is active when cap-dependent translation initiation is inhibited. J. Virol. 2020, 95, e01998-20. [Google Scholar] [CrossRef]
- Chen, C.-K.; Cheng, R.; Demeter, J.; Chen, J.; Weingarten-Gabbay, S.; Jiang, L.; Snyder, M.P.; Weissman, J.S.; Segal, E.; Jackson, P.K.; et al. Structured elements drive extensive circular RNA translation. Mol. Cell 2021, 81, 4300–4318. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Starck, S.R.; Shastri, N. Presentation of Cryptic Peptides by MHC Class I Is Enhanced by Inflammatory Stimuli. J. Immunol. 2016, 197, 2981–2991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starck, S.R.; Shastri, N. Nowhere to hide: Unconventional translation yields cryptic peptides for immune surveillance. Immunol. Rev. 2016, 272, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sehta, P.; Wilhelm, A.-M.; Lin, S.-J.; Urman, M.A.; MacNeil, H.A.; Fuchs, G. A Split NanoLuc Reporter Quantitatively Measures Circular RNA IRES Translation. Genes 2022, 13, 357. https://doi.org/10.3390/genes13020357
Sehta P, Wilhelm A-M, Lin S-J, Urman MA, MacNeil HA, Fuchs G. A Split NanoLuc Reporter Quantitatively Measures Circular RNA IRES Translation. Genes. 2022; 13(2):357. https://doi.org/10.3390/genes13020357
Chicago/Turabian StyleSehta, Priyanka, Ann-Marie Wilhelm, Shu-Jun Lin, Michelle A. Urman, Haley A. MacNeil, and Gabriele Fuchs. 2022. "A Split NanoLuc Reporter Quantitatively Measures Circular RNA IRES Translation" Genes 13, no. 2: 357. https://doi.org/10.3390/genes13020357