
The Role of TRPM4 Gene Mutations in Causing Familial Progressive Cardiac Conduction Disease: A Further Contribution

 ,
,
Abstract
:1. Introduction
2. Patients and Methods
Mutation Screening
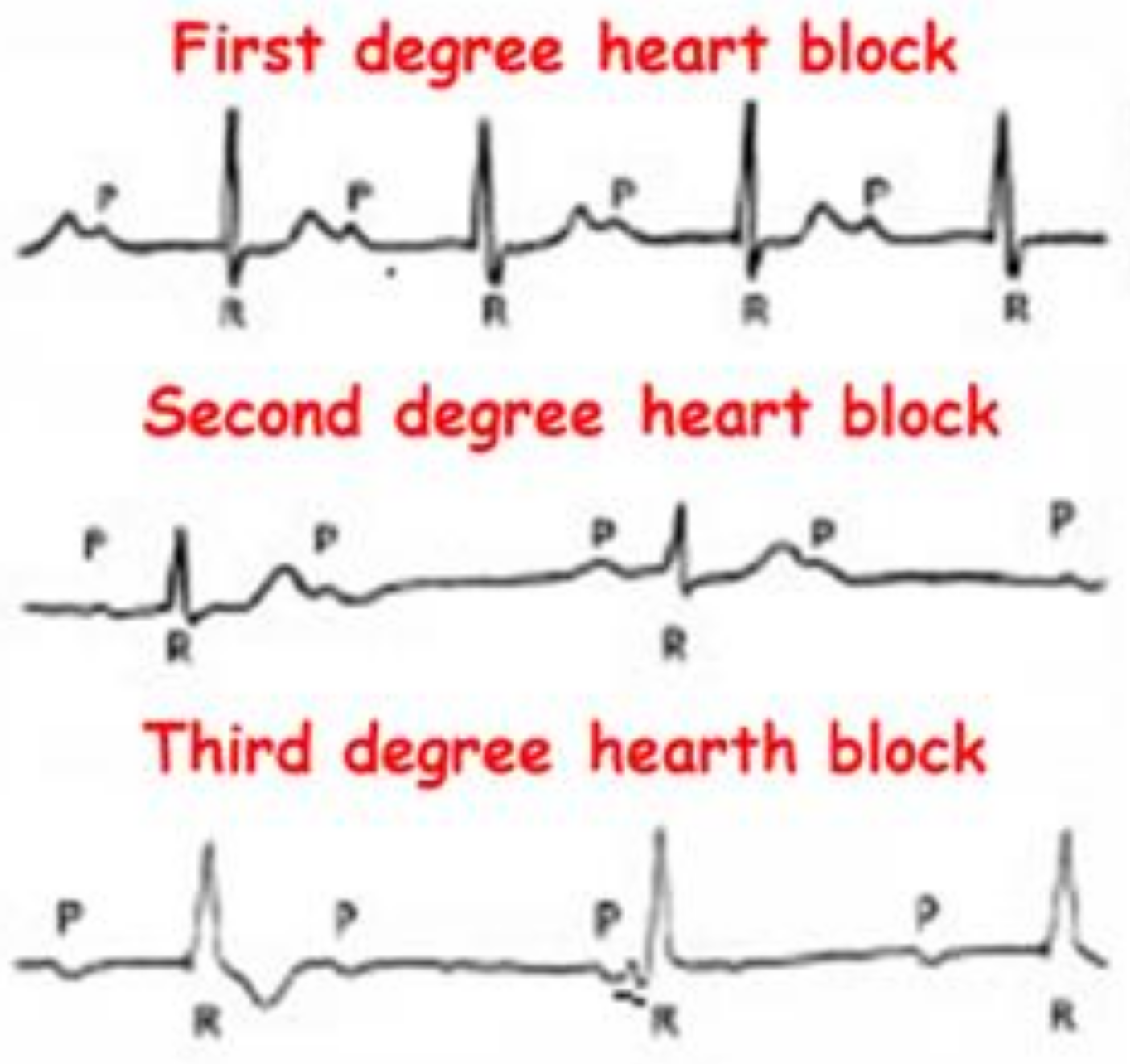
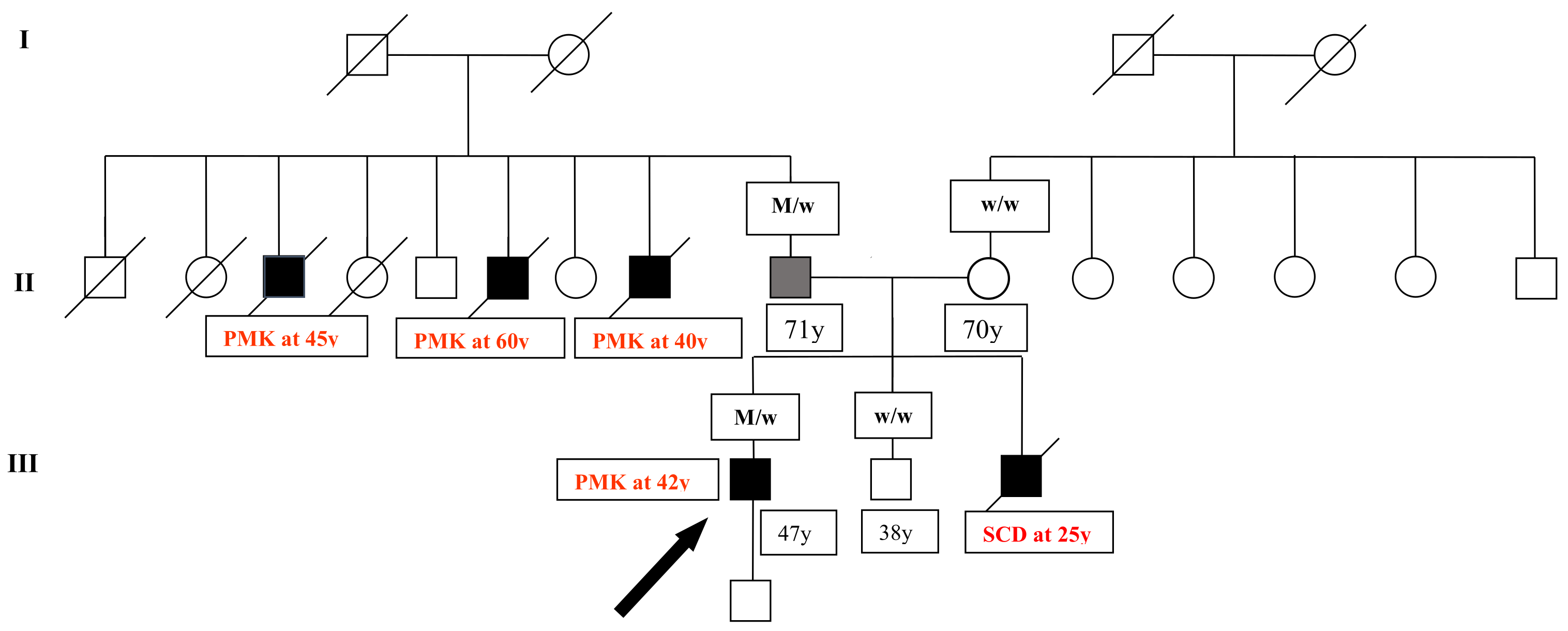
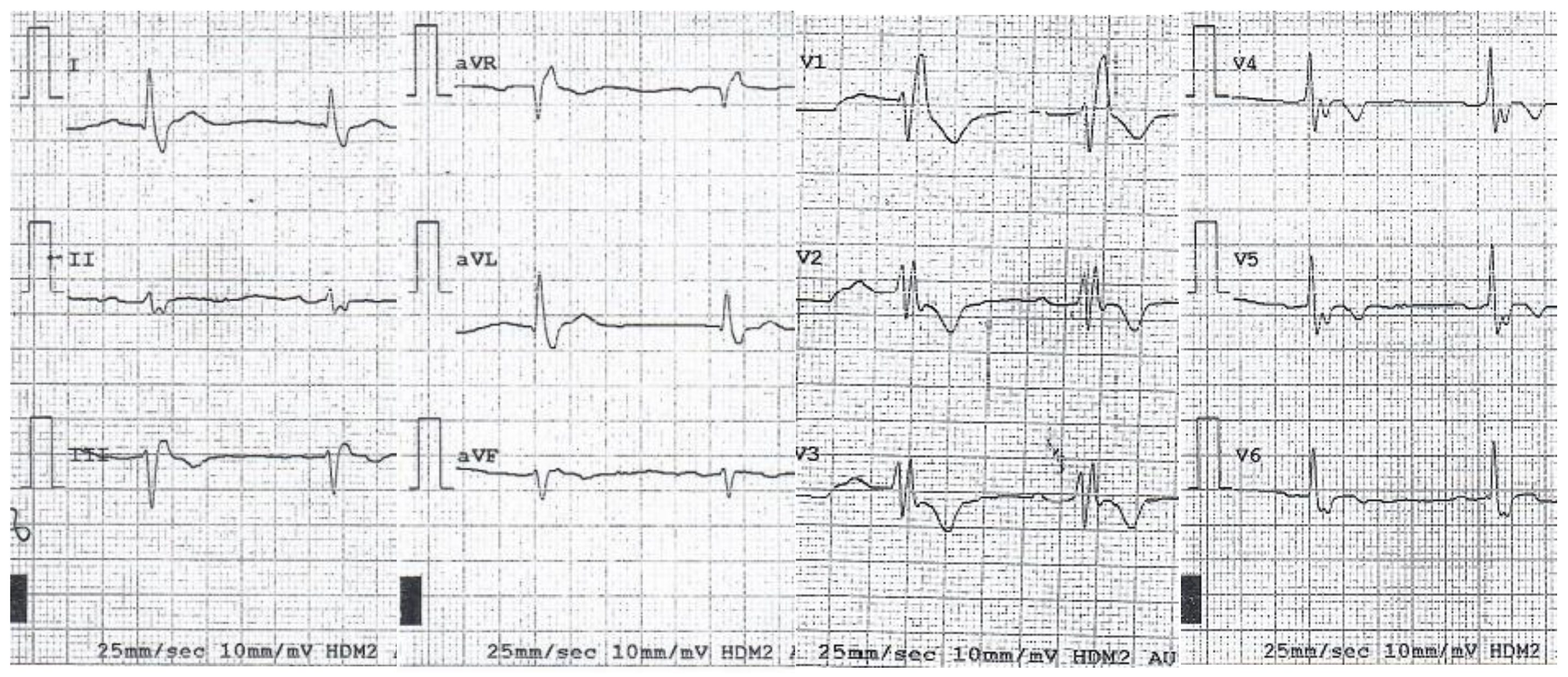
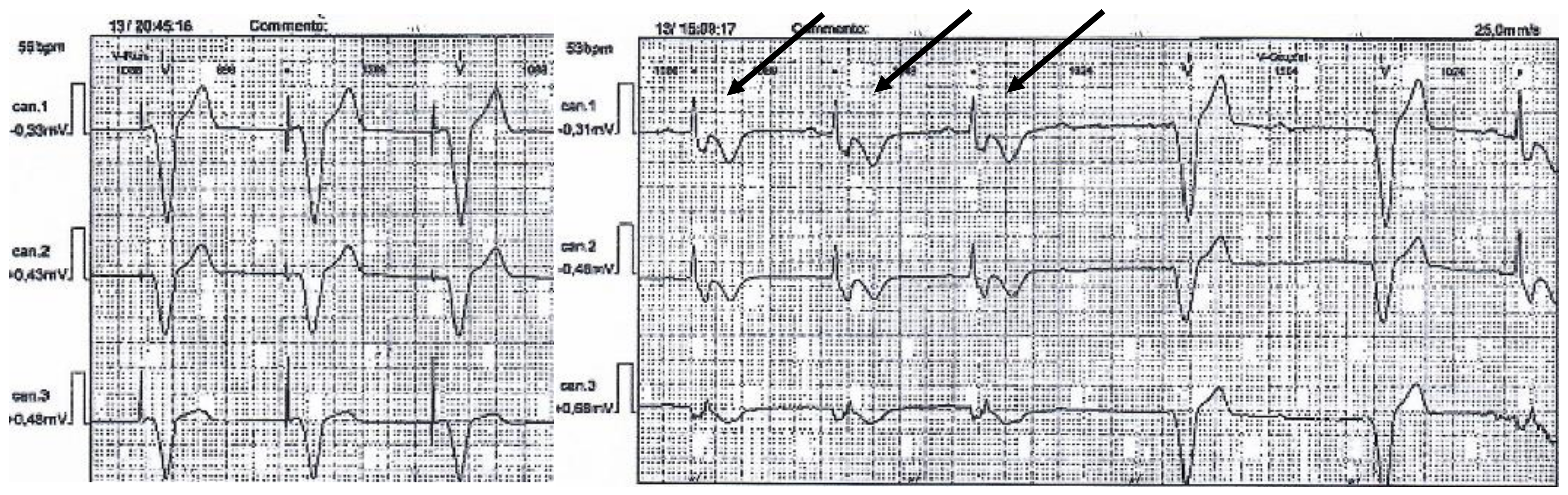
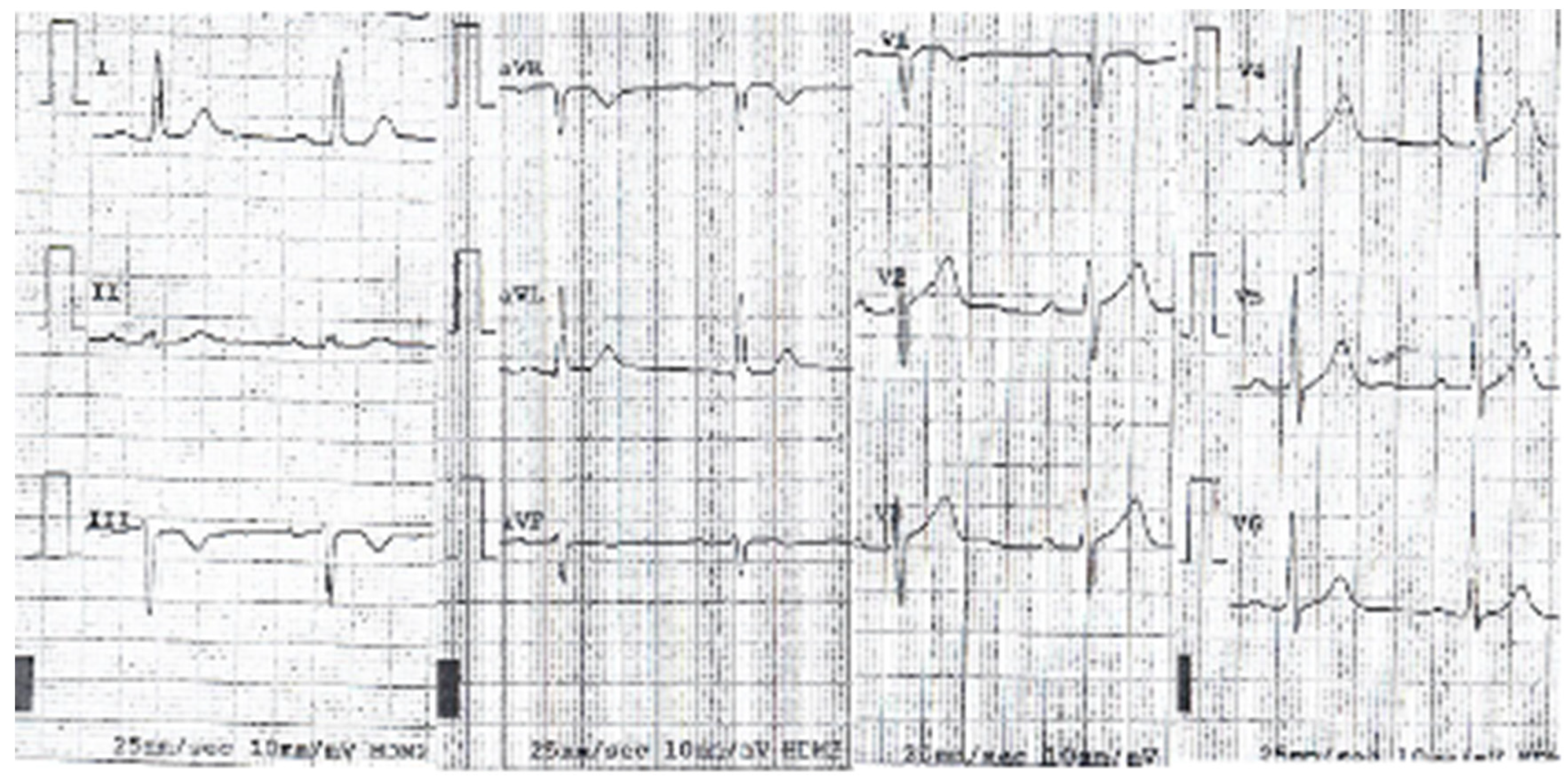
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ector, H.; Rickards, A.F.; Kappenberger, L.; Vardas, P.; Oto, A.; Santini, M.; Sutton, R.; European Working Group on Cardiac Pacing. The registry of the European Working Group on Cardiac Pacing (EWGCP). A working group of the European Society of Cardiology. Europace 2000, 2, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.E.; Di Marco, J.P.; Ellenbogen, K.A.; Estes, N.A., 3rd; Freedman, R.A.; Gettes, L.S.; Gillinov, A.M.; Gregoratos, G.; Hammill, S.C.; Hayes, D.L.; et al. ACC/AHA/HRS 2008 Guidelines for Device-Based Therapy of Cardiac Rhythm Abnormalities: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices): Developed in collaboration with the American Association for Thoracic Surgery and Society of Thoracic Surgeons. Circulation 2008, 117, e350–e408. [Google Scholar] [CrossRef] [Green Version]
- Benson, D.W. Genetics of atrioventricular conduction disease in humans. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2004, 280, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.F.; Yang, J.F.; Wang, Q.; Li, R.G.; Xu, Y.J.; Qu, X.K.; Fang, W.Y.; Liu, X.; Yang, Y.Q. Prevalence and spectrum of GJA5 mutations associated with lone atrial fibrillation. Mol. Med. Rep. 2013, 7, 767–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anselme, F.; Moubarak, G.; Savouré, A.; Godin, B.; Borz, B.; Drouin-Garraud, V.; Gay, A. Implantable cardioverter-defibrillators in lamin A/C mutation carriers with cardiac conduction disorders. Heart Rhythm 2013, 10, 1492–1498. [Google Scholar] [CrossRef]
- Baruteau, A.E.; Probst, V.; Abriel, H. Inherited progressive cardiac conduction disorders. Curr. Opin. Cardiol. 2015, 30, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, R.M.F.L.; de Souza Maciel, A. Conduction Disorders: The Value of Surface ECG. Curr. Cardiol. Rev. 2021, 17, 173–181. [Google Scholar] [CrossRef]
- Brink, A.J.; Torrington, M. Progressive familial heart block-two types. S. Afr. Med. J. 1977, 52, 53–59. [Google Scholar]
- Brink, P.A.; Ferreira, A.; Moolman, J.C.; Weymar, H.W.; van der Merwe, P.L.; Corfield, V.A. Gene for progressive familial heart block type I maps to chromosome 19q13. Circulation 1995, 91, 1633–1640. [Google Scholar] [CrossRef]
- Schott, J.J.; Alshinawi, C.; Kyndt, F.; Probst, V.; Hoorntje, T.M.; Hulsbeek, M.; Wilde, A.A.; Escande, D.; Mannens, M.M.; Le Marec, H. Cardiac conduction defects associate with mutations in SCN5A. Nat. Genet. 1999, 23, 20–21. [Google Scholar] [CrossRef]
- Kruse, M.; Schulze-Bahr, E.; Corfield, V.; Beckmann, A.; Stallmeyer, B.; Kurtbay, G.; Ohmert, I.; Schulze-Bahr, E.; Brink, P.; Pongs, O. Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J. Clin. Investig. 2009, 119, 2737–2744. [Google Scholar] [CrossRef] [PubMed]
- Launay, P.; Fleig, A.; Perraud, A.L.; Scharenberg, A.M.; Penner, R.; Kinet, J.P. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell 2002, 109, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Nilius, B.; Prenen, J.; Droogmans, G.; Voets, T.; Vennekens, R.; Freichel, M.; Wissenbach, U.; Flockerzi, V. Voltage dependence of the Ca2+-activated cation channel TRPM4. J. Biol. Chem. 2003, 278, 30813–30820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abriel, H.; Syam, N.; Sottas, V.; Amarouch, M.Y.; Rougier, J.S. TRPM4 channels in the cardiovascular system: Physiology, pathophysiology, and pharmacology. Biochem. Pharmacol. 2012, 84, 873–881. [Google Scholar] [CrossRef]
- Kruse, M.; Pongs, O. TRPM4 channels in the cardiovascular system. Curr. Opin. Pharmacol. 2014, 15, 68–73. [Google Scholar] [CrossRef]
- Guinamard, R.; Demion, M.; Chatelier, A.; Bois, P. Calcium-activated nonselective cation channels in mammalian cardiomyocytes. Trends Cardiovasc. Med. 2006, 16, 245–250. [Google Scholar] [CrossRef]
- Guinamard, R.; Chatelier, A.; Demion, M.; Potreau, D.; Patri, S.; Rahmati, M.; Bois, P. Functional characterization of a Ca(2+)-activated non-selective cation channel in human atrial cardiomyocytes. J. Physiol. 2004, 558, 75–83. [Google Scholar] [CrossRef]
- Gonzales, A.L.; Garcia, Z.I.; Amberg, G.C.; Earley, S. Pharmacological inhibition of TRPM4 hyperpolarizes vascular smooth muscle. Am. J. Physiol. Cell Physiol. 2010, 299, C1195–C1202. [Google Scholar] [CrossRef] [Green Version]
- Demion, M.; Thireau, J.; Gueffier, M.; Finan, A.; Khoueiry, Z.; Cassan, C.; Serafini, N.; Aimond, F.; Granier, M.; Pasquié, J.-L.; et al. Trpm4 gene invalidation leads to cardiac hypertrophy and electrophysiological alterations. PLoS ONE 2014, 9, e115256. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, G.; Demydenko, K.; Dries, E.; Puertas, R.D.; Jin, X.; Sipido, K.; Roderick, H.L. Calcium Signaling in Cardiomyocyte Function. Cold Spring Harb. Perspect. Biol. 2020, 12, a035428. [Google Scholar] [CrossRef]
- Wagner, S.; Maier, L.S.; Bers, D.M. Role of sodium and calcium dysregulation in tachy-arrhythmias in sudden cardiac death. Circ. Res. 2015, 116, 1956–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, S.P.T.; Agrawal, S.; Gong, M.; Zhao, J. Modulated Calcium Homeostasis and Release Events Under Atrial Fibrillation and Its Risk Factors: A Meta-Analysis. Front. Cardiovasc. Med. 2021, 8, 662914. [Google Scholar] [CrossRef] [PubMed]
- Guinamard, R.; Bouvagnet, P.; Hof, T.; Liu, H.; Simard, C.; Sallé, L. TRPM4 in cardiac electrical activity. Cardiovasc. Res. 2015, 108, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathar, I.; Jacobs, G.; Kecskes, M.; Menigoz, A.; Philippaert, K.; Vennekens, R. TRPM4. Handb. Exp. Pharmacol. 2014, 222, 461–487. [Google Scholar] [CrossRef]
- Jacobs, G.; Oosterlinck, W.; Dresselaers, T.; Geenens, R.; Kerselaers, S.; Himmelreich, U.; Herijgers, P.; Vennekens, R. Enhanced?-adrenergic cardiac reserve in Trpm4?/? mice with ischaemic heart failure. Cardiovasc. Res. 2015, 105, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Du, R.; Fan, L.L.; Jin, J.Y.; Huang, H.; Chen, Y.Q.; Bi, D.D.; Xiang, R. Whole-Exome Sequencing Identifies a Novel TRPM4 Mutation in a Chinese Family with Atrioventricular Block. Biomed. Res. Int. 2021, 2021, 9247541. [Google Scholar] [CrossRef]
- Daumy, X.; Amarouch, M.Y.; Lindenbaum, P.; Bonnaud, S.; Charpentier, E.; Bianchi, B.; Nafzger, S.; Baron, E.; Fouchard, S.; Thollet, A.; et al. Targeted resequencing identifies TRPM4 as a major gene predisposing to progressive familial heart block type I. Int. J. Cardiol. 2016, 207, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; El Zein, L.; Kruse, M.; Guinamard, R.; Beckmann, A.; Bozio, A.; Kurtbay, G.; Mégarbané, A.; Ohmert, I.; Blaysat, G.; et al. Gain-of-function mutations in TRPM4 cause autosomal dominant isolated cardiac conduction disease. Circ. Cardiovasc. Genet. 2010, 3, 374–385. [Google Scholar] [CrossRef] [Green Version]
- Stallmeyer, B.; Zumhagen, S.; Denjoy, I.; Duthoit, G.; Hébert, J.L.; Ferrer, X.; Maugenre, S.; Schmitz, W.; Kirchhefer, U.; Schulze-Bahr, E.; et al. Mutational spectrum in the Ca(2+)-activated cation channel gene TRPM4 in patients with cardiac conductance disturbances. Hum. Mutat. 2012, 33, 109–117. [Google Scholar] [CrossRef]
- Liu, H.; Chatel, S.; Simard, C.; Syam, N.; Salle, L.; Probst, V.; Morel, J.; Millat, G.; Lopez, M.; Abriel, H.; et al. Molecular genetics and functional anomalies in a series of 248 Brugada cases with 11 mutations in the TRPM4 channel. PLoS ONE 2013, 8, e54131. [Google Scholar] [CrossRef] [Green Version]
- Janin, A.; Bessière, F.; Georgescu, T.; Chanavat, V.; Chevalier, P.; Millat, G. TRPM4 mutations to cause autosomal recessive and not autosomal dominant Brugada type 1 syndrome. Eur. J. Med. Genet. 2019, 62, 103527. [Google Scholar] [CrossRef] [PubMed]
- Hof, T.; Liu, H.; Sallé, L.; Schott, J.J.; Ducreux, C.; Millat, G.; Chevalier, P.; Probst, V.; Guinamard, R.; Bouvagnet, P. TRPM4 non-selective cation channel variants in long QT syndrome. BMC Med. Genet. 2017, 18, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertz, C.L.; Christiansen, S.L.; Larsen, M.K.; Dahl, M.; Ferrero-Miliani, L.; Weeke, P.E.; Pedersen, O.; Hansen, T.; Grarup, N.; Ottesen, G.L.; et al. Genetic investigations of sudden unexpected deaths in infancy using next-generation sequencing of 100 genes associated with cardiac diseases. Eur. J. Hum. Genet. 2016, 24, 817–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, B.; Ozhathil, L.C.; Medeiros-Domingo, A.; Gollob, M.H.; Abriel, H. Four TRPM4 Cation Channel Mutations Found in Cardiac Conduction Diseases Lead to Altered Protein Stability. Front Physiol. 2018, 9, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarouch, M.Y.; El Hilaly, J. Inherited Cardiac Arrhythmia Syndromes: Focus on Molecular Mechanisms Underlying TRPM4 Channelopathies. Cardiovasc. Ther. 2020, 2020, 6615038. [Google Scholar] [CrossRef]
- Celestino-Soper, P.B.; Doytchinova, A.; Steiner, H.A.; Uradu, A.; Lynnes, T.C.; Groh, W.J.; Miller, J.M.; Lin, H.; Gao, H.; Wang, Z.; et al. Evaluation of the Genetic Basis of Familial Aggregation of Pacemaker Implantation by a Large Next Generation Sequencing Panel. PLoS ONE 2015, 10, e0143588. [Google Scholar] [CrossRef] [Green Version]
- Syam, N.; Chatel, S.; Ozhathil, L.C.; Sottas, V.; Rougier, J.S.; Baruteau, A.; Baron, E.; Amarouch, M.Y.; Daumy, X.; Probst, V.; et al. Variants of Transient Receptor Potential Melastatin Member 4 in Childhood Atrioventricular Block. J. Am. Heart Assoc. 2016, 5, e001625. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype | Codon Change | Amino Acid Change | Protein | Reference of First Description |
|---|---|---|---|---|
| Brugada Syndrome | GGG-AGG | Gly-Arg | Gly555Arg | Liu, 2013 [30] |
| TTC-ATC | Phe-Ile | Phe773Ile | Liu, 2013 [30] | |
| CCG-CGG | Pro-Arg | Pro779Arg | Liu, 2013 [30] | |
| CAG-CGG | Gln-Arg | Gln854Arg | Liu, 2013 [30] | |
| ACC-ATC | Thr-Ile | Thr873Ile | Liu, 2013 [30] | |
| AAA-TAA | Lys-Term | Lys914Term | Liu, 2013 [30] | |
| CTG-CCG | Leu-Pro | Leu1075Pro | Liu, 2013 [30] | |
| CCG-CTG | Pro-Leu | Pro1204Leu | Liu, 2013 [30] | |
| Brugada Syndrome (?) | CGG-TGG | Arg-Trp | Arg144Trp | Liu, 2013 [30] |
| Cardiac Conduction Disease | CAG-CAC | Gln-His | Gln131His | Stallmeyer, 2012 [29] |
| CGG-TGG | Arg-Trp | Arg164Trp | Liu, 2013 [30] | |
| CAG-CGG | Gln-Arg | Gln293Arg | Stallmeyer, 2012 [29] | |
| GCC-ACC | Ala-Thr | Ala432Thr | Liu, 2010 [28] | |
| GGT-AGT | Gly-Ser | Gly582Ser | Stallmeyer, 2012 [29] | |
| TAC-CAC | Tyr-His | Tyr790His | Stallmeyer, 2012 [29] | |
| GGC-GAC | Gly-Asp | Gly844Asp | Liu, 2010 [28] | |
| AAA-AGA | Lys-Arg | Lys914Arg | Stallmeyer, 2012 [29] | |
| CCC-TCC | Pro-Ser | Pro970Ser | Stallmeyer, 2012 [29] | |
| Heart Block Type 1 | GAG-AAG | Glu- Lys | Glu7Lys | Kruse, 2009 [11] |
| ATA- ACA | Ile-Thr | Ile376Thr | Daumy, 2016 [27] | |
| Long QT Syndrome | CTG-ATG | Val-Met | Val441Met | Hof, 2017 [32] |
| CGG-CCG | Arg-Pro | Arg499Pro | Hof, 2017 [32] | |
| CGG-TGG | Arg-Trp | Arg499Trp | Hof, 2017 [32] | |
| Sudden unexpected death in infancy | TGG-TGA | Trp-Term | Trp5252Term | Hertz, 2016 [33] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palladino, A.; Papa, A.A.; Petillo, R.; Scutifero, M.; Morra, S.; Passamano, L.; Nigro, V.; Politano, L. The Role of TRPM4 Gene Mutations in Causing Familial Progressive Cardiac Conduction Disease: A Further Contribution. Genes 2022, 13, 258. https://doi.org/10.3390/genes13020258
Palladino A, Papa AA, Petillo R, Scutifero M, Morra S, Passamano L, Nigro V, Politano L. The Role of TRPM4 Gene Mutations in Causing Familial Progressive Cardiac Conduction Disease: A Further Contribution. Genes. 2022; 13(2):258. https://doi.org/10.3390/genes13020258
Chicago/Turabian StylePalladino, Alberto, Andrea Antonio Papa, Roberta Petillo, Marianna Scutifero, Salvatore Morra, Luigia Passamano, Vincenzo Nigro, and Luisa Politano. 2022. "The Role of TRPM4 Gene Mutations in Causing Familial Progressive Cardiac Conduction Disease: A Further Contribution" Genes 13, no. 2: 258. https://doi.org/10.3390/genes13020258