
Combining Fine Mapping, Whole-Genome Re-Sequencing, and RNA-Seq Unravels Candidate Genes for a Soybean Mutant with Short Petioles and Weakened Pulvini


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
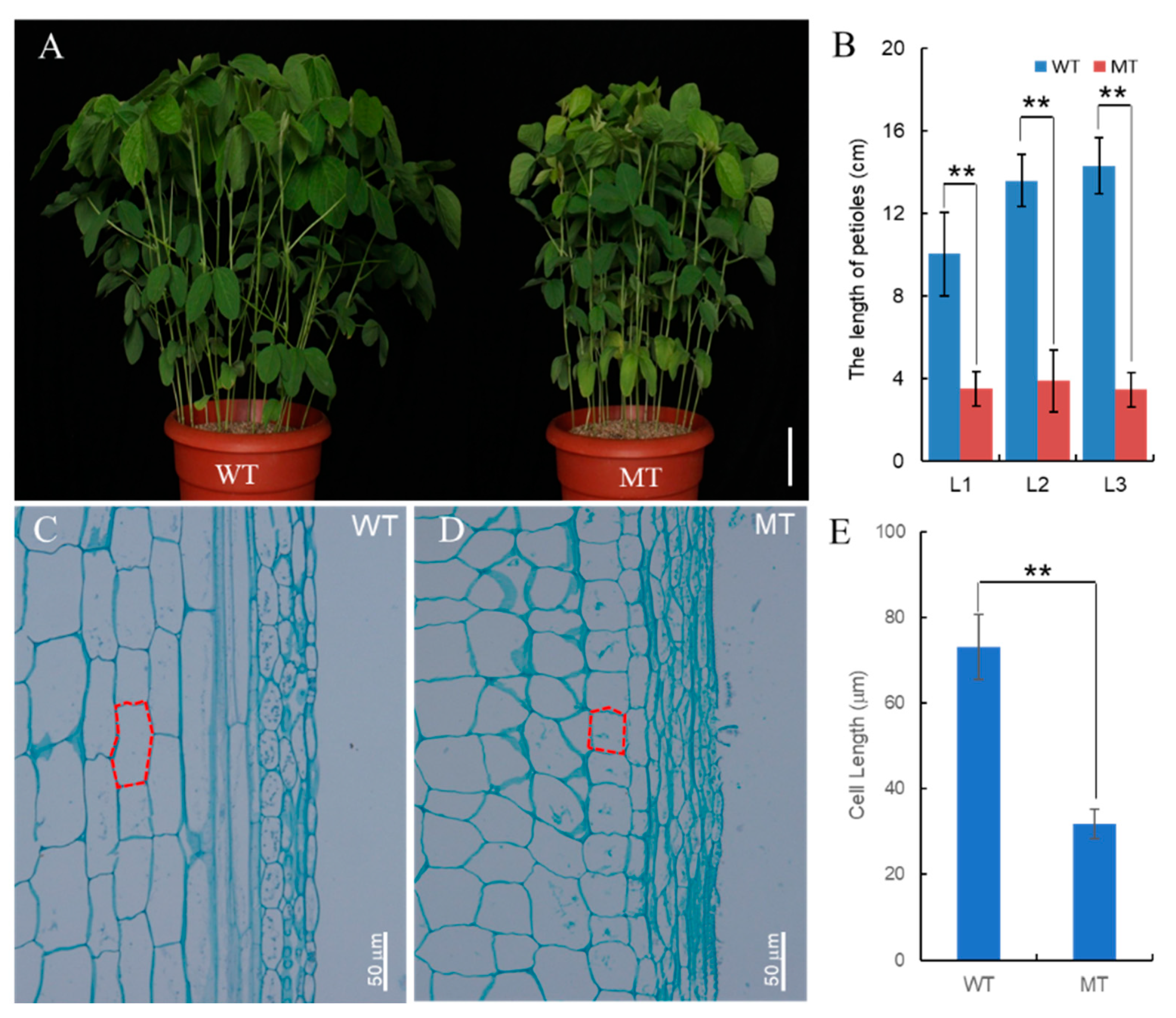
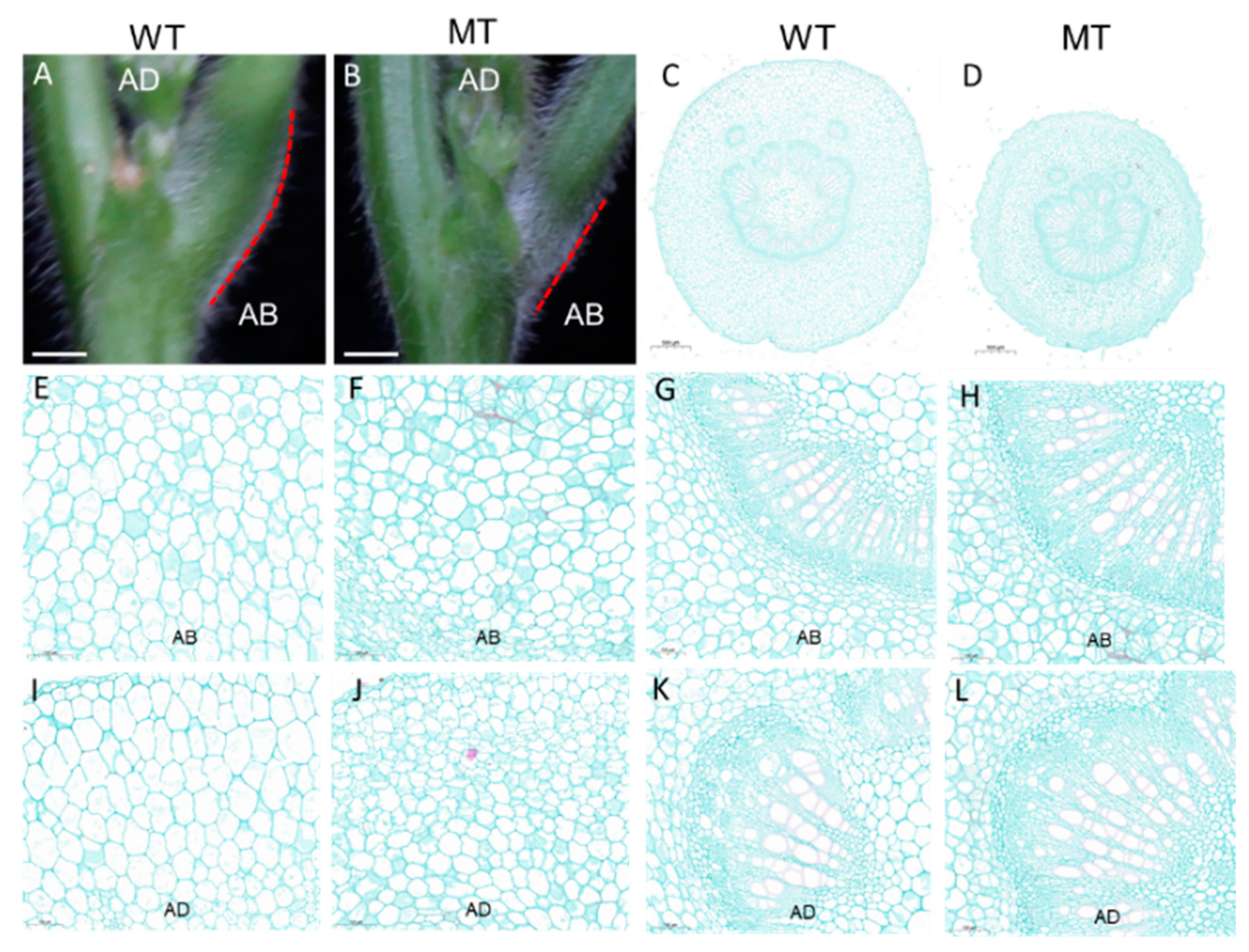
2.2. Histological Analysis
2.3. Inheritance Analysis and Molecular Mapping with Simple Sequence Repeat (SSR) Markers
2.4. DNA Library Construction and Whole-Genome Re-Sequencing
2.5. RNA Isolation and Quantitative Real-Time RT-PCR Analysis
2.6. RNA-Seq analysis
3. Results
3.1. Phenotypic Characterization of the Spwp Mutant
3.2. Genetic Analysis of the Spwp Mutant
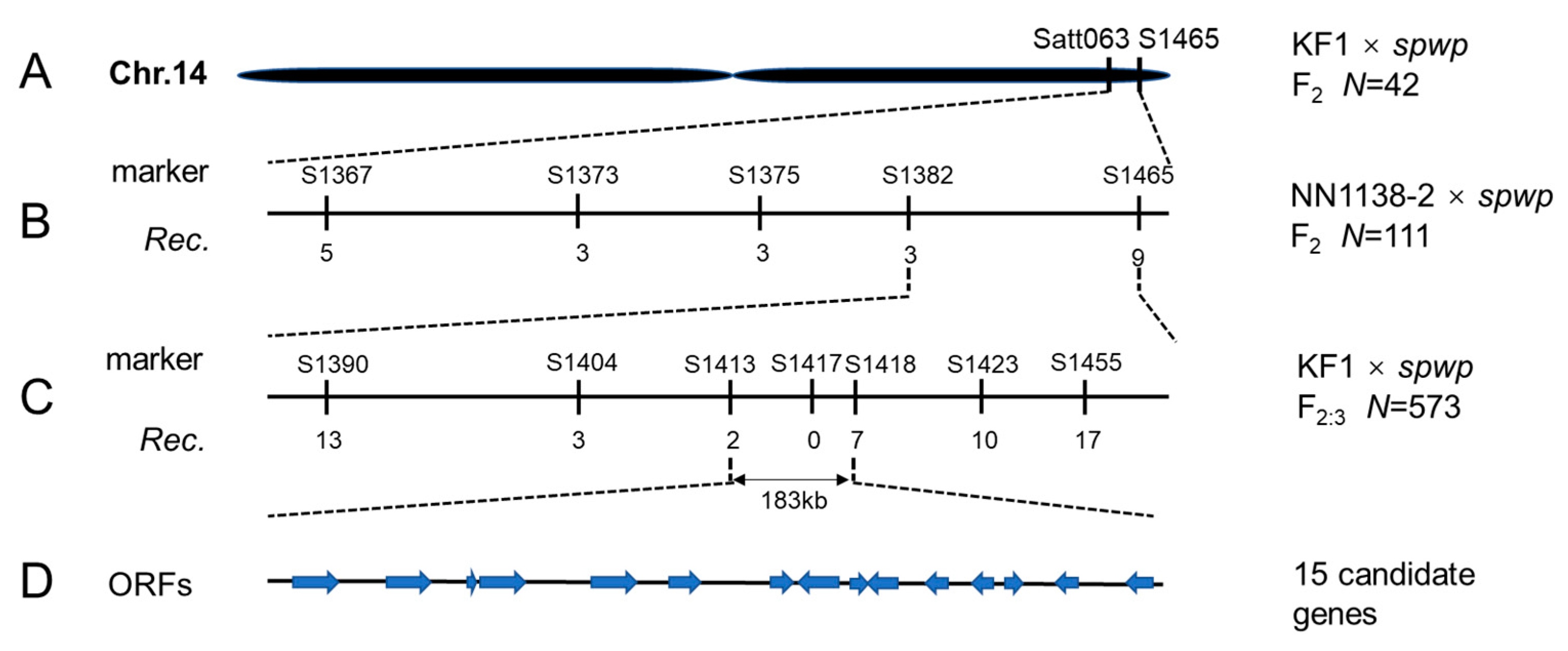
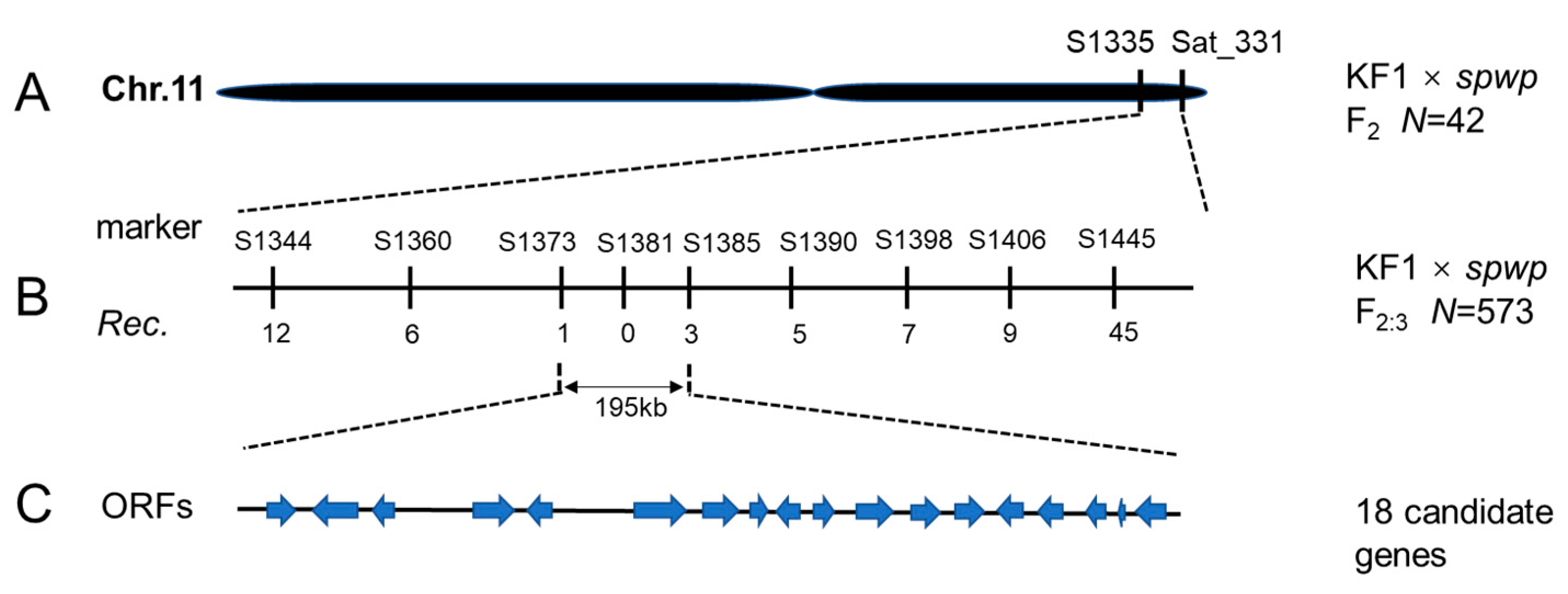
3.3. Fine Mapping of the Spwp1 and Spwp2 Genes
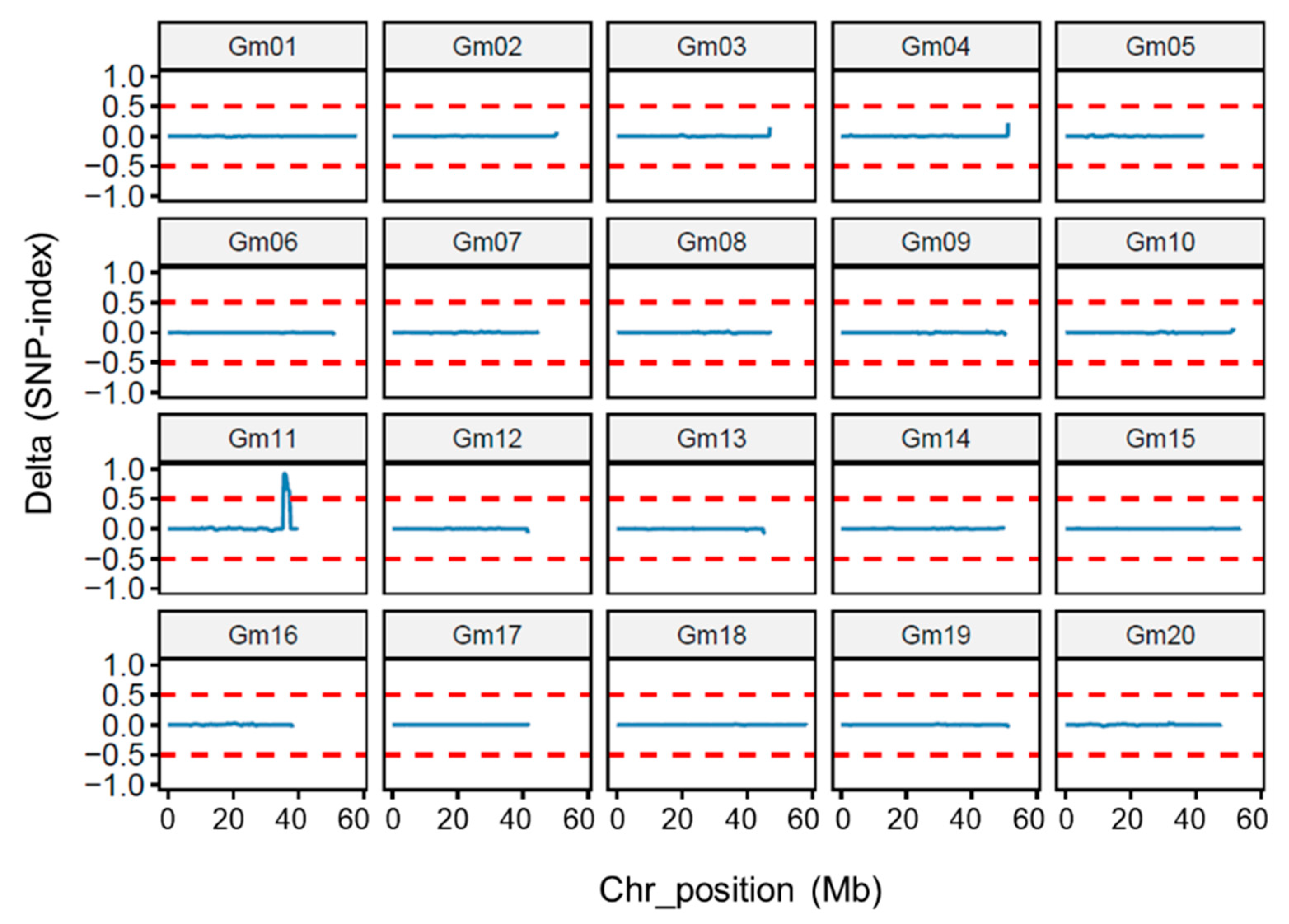
3.4. Whole-Genome Re-Sequencing Analysis
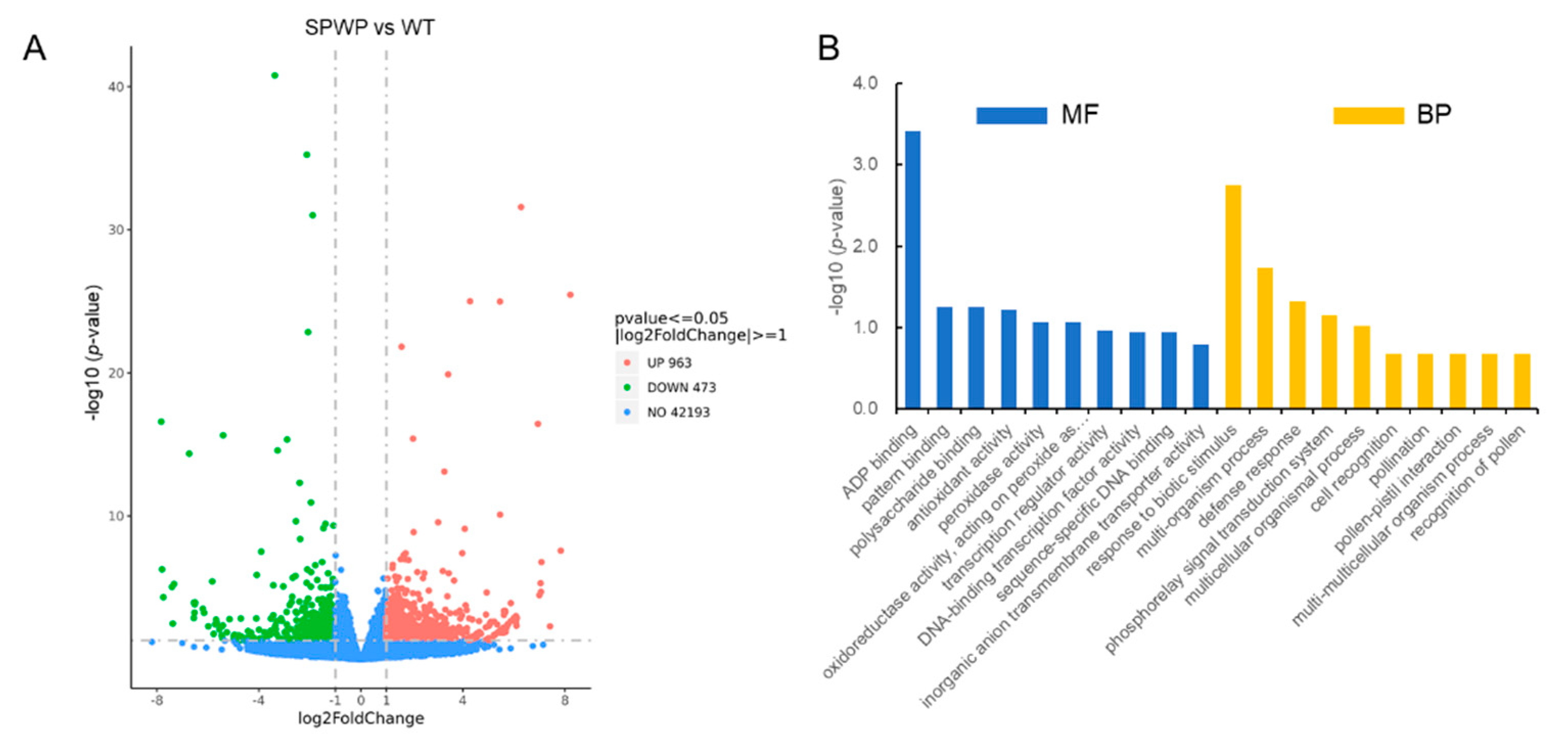
3.5. RNA-Seq Analysis between the Spwp Mutant and WT
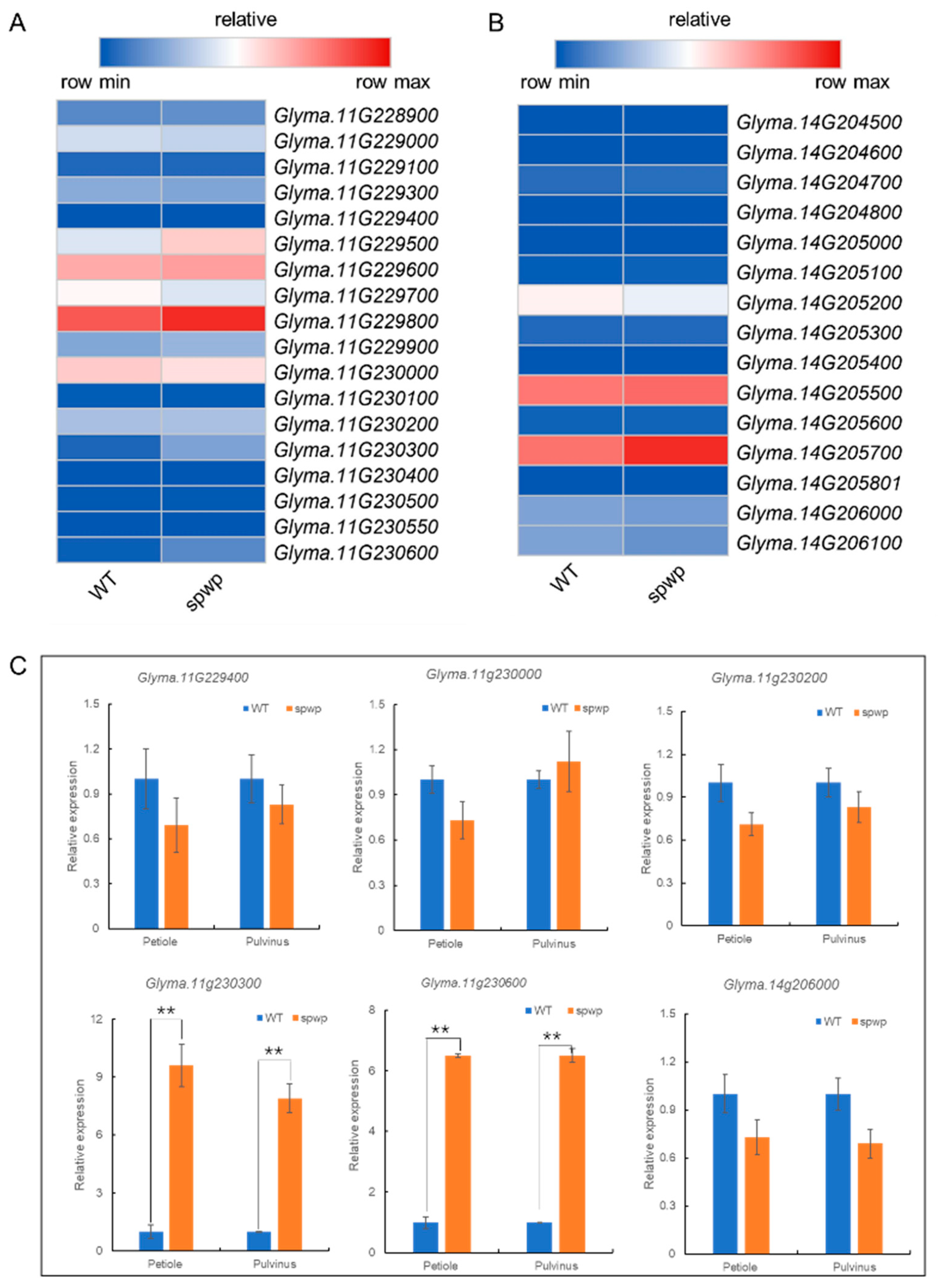
3.6. Candidate Genes Analysis by Integrating Whole-Genome Re-Sequencing and RNA-Seq Data
3.7. Enrichment Analysis on DEGs Associated with the Phenotype of the Spwp Mutant
4. Discussion
4.1. Spwp Is a New Soybean Short Petiole Mutant with Weakened Pulvini
4.2. Identification Candidate Genes of the Spwp Mutant through Whole-Genome Re-Sequencing and RNA-Seq
4.3. The Spwp Mutant Also Showed Defective Leaf Movement
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| RNA-seq | RNA sequencing |
| AUX/IAA | Auxin/indoleacetic acid |
| SAUR | SMALL AUXIN-UP RNA |
| PIN3A | PIN-FORMED 3A |
| PIN3B | PIN-FORMED 3B |
| bHLH | Basic helix–loop–helix protein |
| WRKY | Transcription factor (TF), partake WRKYGQK heptapeptide at N-terminal |
| MYB | Myeloblastosis (avian myeloblastosis viral oncogene homolog) TF |
| ZF | Zinc finger TF |
| AP2/EREBP | Apetala2/ethylene-responsive element binding protein |
| NB-ARC | Nucleotide-binding domain shared with APAF-1, various R proteins, and CED-4 |
| CLE40 | CLV3/EMBRYO SURROUNDING REGION (ESR) 40 |
| WOX5 | WUSCHEL-RELATED HOMEOBOX5 |
References
- Godin, C. Representing and encoding plant architecture: A review. Ann. Forest Sci. 2000, 57, 413–438. [Google Scholar] [CrossRef]
- Kilen, T. Inheritance of a Short Petiole Trait in Soybean 1. Crop Sci. 1983, 23, 1208–1210. [Google Scholar] [CrossRef]
- You, M.G.; Zhao, T.J.; Gai, J.Y.; Yen, Y. Genetic analysis of short petiole and abnormal pulvinus in soybean. Euphytica 1998, 102, 329–333. [Google Scholar] [CrossRef]
- Jun, T.-H.; Kang, S.-T.; Moon, J.-K.; Seo, M.-J.; Yun, H.-T.; Lee, S.-K.; Lee, Y.-H.; Kim, S.-J. Genetic analysis of new short petiole gene in soybean. J. Crop Sci. Biotechnol. 2009, 12, 87–89. [Google Scholar] [CrossRef]
- Chen, L.M.; Yang, H.L.; Fang, Y.S.; Guo, W.; Chen, H.F.; Zhang, X.J.; Dai, W.J.; Chen, S.L.; Hao, Q.N.; Yuan, S.L.; et al. Overexpression of GmMYB14 improves high-density yield and drought tolerance of soybean through regulating plant architecture mediated by the brassinosteroid pathway. Plant Biotechnol. J. 2021, 19, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Van der Graaff, E.; Den Dulk-Ras, A.; Hooykaas, P.J.J.; Keller, B. Activation tagging of the LEAFY PETIOLE gene affects leaf petiole development in Arabidopsis thaliana. Development 2000, 127, 4971–4980. [Google Scholar] [CrossRef] [PubMed]
- Ha, C.M.; Kim, G.T.; Kim, B.C.; Jun, J.H.; Soh, M.S.; Ueno, Y.; Machida, Y.; Tsukaya, H.; Nam, H.G. The BLADE-ON-PETIOLE 1 gene controls leaf pattern formation through the modulation of meristematic activity in Arabidopsis. Development 2003, 130, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Ha, C.M.; Jun, J.H.; Nam, H.G.; Fletcher, J.C. BLADE-ON-PETIOLE1 encodes a BTB/POZ domain protein required for leaf morphogenesis in Arabidopsis thaliana. Plant Cell Physiol. 2004, 45, 1361–1370. [Google Scholar] [CrossRef]
- Dinneny, J.R.; Yadegari, R.; Fischer, R.L.; Yanofsky, M.F.; Weigel, D. The role of JAGGED in shaping lateral organs. Development 2004, 131, 1101–1110. [Google Scholar] [CrossRef] [Green Version]
- Ohno, C.K.; Reddy, G.V.; Heisler, M.G.B.; Meyerowitz, E.M. The Arabidopsis JAGGED gene encodes a zinc finger protein that promotes leaf tissue development. Development 2004, 131, 1111–1122. [Google Scholar] [CrossRef] [Green Version]
- Favero, D.S.; Kawamura, A.; Shibata, M.; Takebayashi, A.; Jung, J.-H.; Suzuki, T.; Jaeger, K.E.; Ishida, T.; Iwase, A.; Wigge, P.A. AT-hook transcription factors restrict petiole growth by antagonizing PIFs. Curr. Biol. 2020, 30, 1454–1466. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, P.J.; Millar, A.J. Biological Rhythms and Photoperiodism in Plants; Bios Scientific Publishers: Oxford, UK, 1998; Volume 82, pp. 900–901. [Google Scholar]
- Mayer, W.-E.; Hampp, R. Movement of pulvinated leaves. In Progress in Botany; Springer: Berlin/Heidelberg, Germany, 1995; pp. 236–262. [Google Scholar]
- Uehlein, N.; Kaldenhoff, R. Aquaporins and plant leaf movements. Ann. Bot. 2008, 101, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, N. Osmoregulation of leaf motor cells. FEBS Lett. 2007, 581, 2337–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortizo, M.; Laufs, P. Genetic basis of the “sleeping leaves” revealed. Proc. Natl. Acad. Sci. USA 2012, 109, 11474–11475. [Google Scholar] [CrossRef] [Green Version]
- Cote, G.G. Signal-Transduction in Leaf Movement. Plant Physiol. 1995, 109, 729–734. [Google Scholar] [CrossRef]
- Moshelion, M.; Becker, D.; Czempinski, K.; Mueller-Roeber, B.; Attali, B.; Hedrich, R.; Moran, N. Diurnal and circadian regulation of putative potassium channels in a leaf moving organ. Plant Physiol. 2002, 128, 634–642. [Google Scholar] [CrossRef]
- Marx, G.A. A suite of mutants that modify pattern formation in pea leaves. Plant Mol. Biol. Rep. 1987, 5, 311–335. [Google Scholar] [CrossRef]
- Kawaguchi, M. SLEEPLESS, a gene conferring nyctinastic movement in legume. J. Plant Res. 2003, 116, 151–154. [Google Scholar] [CrossRef]
- Zhou, C.N.; Han, L.; Fu, C.X.; Chai, M.F.; Zhang, W.Z.; Li, G.F.; Tang, Y.H.; Wang, Z.Y. Identification and characterization of petiolule- like pulvinus mutants with abolished nyctinastic leaf movement in the model legume Medicago truncatula. New Phytol. 2012, 196, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.H.; Moreau, C.; Liu, Y.; Kawaguchi, M.; Hofer, J.; Ellis, N.; Chen, R.J. Conserved genetic determinant of motor organ identity in Medicago truncatula and related legumes. Proc. Natl. Acad. Sci. USA 2012, 109, 11723–11728. [Google Scholar] [CrossRef] [Green Version]
- Shuai, B.; Reynaga-Pena, C.G.; Springer, P.S. The lateral organ boundaries gene defines a novel, plant-specific gene family. Plant Physiol. 2002, 129, 747–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.S.; Yang, S.X.; Cheng, W.; Fu, Y.F.; Leng, J.T.; Yuan, X.H.; Jiang, N.; Ma, J.X.; Feng, X.Z. GmILPA1, Encoding an APC8-like Protein, Controls Leaf Petiole Angle in Soybean. Plant Physiol. 2017, 174, 1167–1176. [Google Scholar] [PubMed] [Green Version]
- You, M.; Zhao, T.; Gai, J. Performance and inheritance of short petiole trait of a new soybean mutant. Soybean Genet. Newsl. 1995, 22, 71–75. [Google Scholar]
- Michelmore, R.W.; Paran, I.; Kesseli, R.V. Identification of Markers Linked to Disease-Resistance Genes by Bulked Segregant Analysis: A Rapid Method to Detect Markers in Specific Genomic Regions by Using Segregating Populations. Proc. Natl. Acad. Sci. USA 1991, 88, 9828–9832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Q.J.; Jia, G.F.; Zhu, Y.L.; Grant, D.; Nelson, R.T.; Hwang, E.Y.; Hyten, D.L.; Cregan, P.B. Abundance of SSR Motifs and Development of Candidate Polymorphic SSR Markers (BARCSOYSSR_1.0) in Soybean. Crop Sci. 2010, 50, 1950–1960. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Volkov, A.G.; Foster, J.C.; Baker, K.D.; Markin, V.S. Mechanical and electrical anisotropy in Mimosa pudica pulvini. Plant Signal. Behav. 2010, 5, 1211–1221. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Yeom, E.; Lee, S.J. Real-time imaging of pulvinus bending in Mimosa pudica. Sci. Rep. 2014, 4, 6466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, I.; Sheen, J. Two-component circuitry in Arabidopsis cytokinin signal transduction. Nature 2001, 413, 383–389. [Google Scholar] [CrossRef] [PubMed]
- To, J.P.C.; Haberer, G.; Ferreira, F.J.; Deruere, J.; Mason, M.G.; Schaller, G.E.; Alonso, J.M.; Ecker, J.R.; Kieber, J.J. Type-A Arabidopsis response regulators are partially redundant negative regulators of cytokinin signaling. Plant Cell 2004, 16, 658–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frebort, I.; Kowalska, M.; Hluska, T.; Frebortova, J.; Galuszka, P. Evolution of cytokinin biosynthesis and degradation. J. Exp. Bot. 2011, 62, 2431–2452. [Google Scholar] [CrossRef]
- Wolters, H.; Jurgens, G. Survival of the flexible: Hormonal growth control and adaptation in plant development. Nat. Rev. Genet. 2009, 10, 305–317. [Google Scholar] [CrossRef]
- Seo, P.J.; Xiang, F.N.; Qiao, M.; Park, J.Y.; Lee, Y.N.; Kim, S.G.; Lee, Y.H.; Park, W.J.; Park, C.M. The MYB96 Transcription Factor Mediates Abscisic Acid Signaling during Drought Stress Response in Arabidopsis. Plant Physiol. 2009, 151, 275–289. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.D.; Lee, E.; Lucas, J.R.; Morohashi, K.; Li, D.M.; Murray, J.A.H.; Sack, F.D.; Grotewold, E. Regulation of Cell Proliferation in the Stomatal Lineage by the Arabidopsis Myb Four Lips via Direct Targeting of Core Cell Cycle Genes. Plant Cell 2010, 22, 2306–2321. [Google Scholar] [CrossRef] [Green Version]
- Duek, P.D.; Fankhauser, C. bHLH class transcription factors take centre stage in phytochrome signalling. Trends Plant Sci. 2005, 10, 51–54. [Google Scholar] [CrossRef]
- Zhao, M.; Morohashi, K.; Hatlestad, G.; Grotewold, E.; Lloyd, A. The TTG1-bHLH-MYB complex controls trichome cell fate and patterning through direct targeting of regulatory loci. Development 2008, 135, 1991–1999. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.Y.; Bai, M.Y.; Wu, J.X.; Zhu, J.Y.; Wang, H.; Zhang, Z.G.; Wang, W.F.; Sun, Y.; Zhao, J.; Sun, X.H.; et al. Antagonistic HLH/bHLH Transcription Factors Mediate Brassinosteroid Regulation of Cell Elongation and Plant Development in Rice and Arabidopsis. Plant Cell 2009, 21, 3767–3780. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhang, J.; Wu, Y.-W.; Wang, Y.; Zhang, J.; Zheng, Y.; Li, Y.; Li, X.-B. bHLH transcription factors LP1 and LP2 regulate longitudinal cell elongation. Plant Physiol. 2021, 187, 2577–2591. [Google Scholar] [CrossRef] [PubMed]
- Jun, T.H.; Kang, S.T. Genetic map of lps3: A new short petiole gene in soybeans. Genome 2012, 55, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Bolon, Y.-T.; Haun, W.J.; Xu, W.W.; Grant, D.; Stacey, M.G.; Nelson, R.T.; Gerhardt, D.J.; Jeddeloh, J.A.; Stacey, G.; Muehlbauer, G.J. Phenotypic and genomic analyses of a fast neutron mutant population resource in soybean. Plant physiol. 2011, 156, 240–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.F.; Wang, Y.Q.; Gai, J.Y.; Bhat, J.A.; Li, Y.W.; Kong, J.J.; Zhao, T.J. Genetic Analysis and Gene Mapping for a Short-Petiole Mutant in Soybean (Glycine max (L.) Merr.). Agronomy 2019, 9, 709. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Liu, C.K.; Tu, B.J.; Li, Y.S.; Chen, H.; Zhang, Q.Y.; Liu, X.B. Characterization on a Novel Rolled Leaves and Short Petioles Soybean Mutant Based on Seq-BSA and RNA-Seq Analysis. J. Plant Biol. 2021, 17, 117. [Google Scholar] [CrossRef]
- Yin, J.L.; Wang, L.Q.; Jin, T.T.; Nie, Y.; Liu, H.; Qiu, Y.L.; Yang, Y.H.; Li, B.W.; Zhang, J.J.; Wang, D.G.; et al. A cell wall-localized NLR confers resistance to Soybean mosaic virus by recognizing viral- encoded cylindrical inclusion protein. Mol. Plant 2021, 14, 1881–1900. [Google Scholar] [CrossRef]
- Becraft, P.W.; Stinard, P.S.; McCarty, D.R. CRINKLY4: A TNFR-like receptor kinase involved in maize epidermal differentiation. Science 1996, 273, 1406–1409. [Google Scholar] [CrossRef]
- Watanabe, M.; Tanaka, H.; Watanabe, D.; Machida, C.; Machida, Y. The ACR4 receptor-like kinase is required for surface formation of epidermis-related tissues in Arabidopsis thaliana. Plant J. 2004, 39, 298–308. [Google Scholar] [CrossRef]
- Gifford, M.L.; Dean, S.; Ingram, G.C. The Arabidopsis ACR4 gene plays a role in cell layer organisation during ovule integument and sepal margin development. Development 2003, 130, 4249–4258. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Watanabe, M.; Watanabe, D.; Tanaka, T.; Machida, C.; Machida, Y. ACR4, a putative receptor kinase gene of Arabidopsis thaliana, that is expressed in the outer cell layers of embryos and plants, is involved in proper embryogenesis. Plant Cell Physiol. 2002, 43, 419–428. [Google Scholar] [CrossRef]
- Pu, C.X.; Ma, Y.; Wang, J.; Zhang, Y.C.; Jiao, X.W.; Hu, Y.H.; Wang, L.L.; Zhu, Z.G.; Sun, D.; Sun, Y. Crinkly4 receptor-like kinase is required to maintain the interlocking of the palea and lemma, and fertility in rice, by promoting epidermal cell differentiation. Plant J. 2012, 70, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Pu, C.-X.; Sun, Y. Rice Crinkly4 receptor-like kinase positively regulates culm elongation and amino acid K532 is not essential for its kinase activity. Plant Signal. Behav. 2012, 7, 1062–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, K.; Sandal, P.; Williams, E.L.; Murphy, E.; Stes, E.; Nikonorova, N.; Ramakrishna, P.; Czyzewicz, N.; Montero-Morales, L.; Kumpf, R.; et al. PP2A-3 interacts with ACR4 and regulates formative cell division in the Arabidopsis root. Proc. Natl. Acad. Sci. USA 2016, 113, 1447–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Smet, I.; Vassileva, V.; De Rybel, B.; Levesque, M.P.; Grunewald, W.; Van Damme, D.; Van Noorden, G.; Naudts, M.; Van Isterdael, G.; De Clercq, R.; et al. Receptor-like kinase ACR4 restricts formative cell divisions in the Arabidopsis root. Science 2008, 322, 594–597. [Google Scholar] [CrossRef] [PubMed]
- Stahl, Y.; Wink, R.H.; Ingram, G.C.; Simon, R. A Signaling Module Controlling the Stem Cell Niche in Arabidopsis Root Meristems. Curr. Biol. 2009, 19, 909–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, Y.; Grabowski, S.; Bleckmann, A.; Kuhnemuth, R.; Weidtkamp-Peters, S.; Pinto, K.G.; Kirschner, G.K.; Schmid, J.B.; Wink, R.H.; Hulsewede, A.; et al. Moderation of Arabidopsis Root Sternness by Clavata1 and Arabidopsis Crinkly4 Receptor Kinase Complexes. Curr. Biol. 2013, 23, 362–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.F.; Hu, C.; Cui, Y.W.; Zeng, L.; Li, S.J.N.; Zhu, M.S.; Meng, F.H.; Huang, S.T.; Long, L.; Yi, J.; et al. Conserved and differentiated functions of CIK receptor kinases in modulating stem cell signaling in Arabidopsis. Mol. Plant 2021, 14, 1119–1134. [Google Scholar] [CrossRef]
- Berckmans, B.; Kirschner, G.; Gerlitz, N.; Stadler, R.; Simon, R. CLE40 Signaling Regulates Root Stem Cell Fate. Plant Physiol. 2020, 182, 1776–1792. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.H.; Liu, Y.B.; Bai, B.; Zhang, X.S. Establishment of embryonic shoot–root axis is involved in auxin and cytokinin response during Arabidopsis somatic embryogenesis. Front. Plant Sci. 2015, 5, 792. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chong, K. The Essential Role of Cytokinin Signaling in Root Apical Meristem Formation during Somatic Embryogenesis. Front. Plant Sci. 2016, 6, 1196. [Google Scholar] [CrossRef] [Green Version]
- Savina, M.S.; Pasternak, T.; Omelyanchuk, N.A.; Novikova, D.D.; Palme, K.; Mironova, V.V.; Lavrekha, V.V. Cell Dynamics in WOX5-Overexpressing Root Tips: The Impact of Local Auxin Biosynthesis. Front. Plant Sci. 2020, 11, 560169. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.L.; Yang, T.Q.; Mao, Y.W.; Liu, Y.; Guo, S.Q.; Wang, R.R.; Fangyue, G.W.; He, L.L.; Zhao, B.L.; Bai, Q.Z.; et al. The F-box protein MIO1/SLB1 regulates organ size and leaf movement in Medicago truncatula. J. Exp. Bot. 2021, 72, 2995–3011. [Google Scholar] [CrossRef] [PubMed]
- Moshelion, M.; Becker, D.; Biela, A.; Uehlein, N.; Hedrich, R.; Otto, B.; Levi, H.; Moran, N.; Kaldenhoff, R. Plasma membrane aquaporins in the motor cells of Samanea saman: Diurnal and circadian regulation. Plant Cell 2002, 14, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, T.; Ishimaru, Y.; Munemasa, S.; Takeuchi, Y.; Washiyama, K.; Hamamoto, S.; Yoshikawa, N.; Mutara, Y.; Uozumi, N.; Ueda, M. Ion Channels Regulate Nyctinastic Leaf Opening in Samanea saman. Curr. Biol. 2018, 28, 2230–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.Y.; Bai, Q.Z.; Zhao, B.L.; Wu, Q.; Wang, C.Q.; Liu, Y.; Yang, T.Q.; Liu, Y.; He, H.; Du, S.S.; et al. The geometry of the compound leaf plays a significant role in the leaf movement of Medicago truncatula modulated by mtdwarf4a. New Phytol. 2021, 230, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.M.; Meng, Z.; Wang, H.F.; Wang, Y.; Zhang, Y.X.; Hong, L.M.; Liu, R.; Wang, M.; Zhang, J.; Han, L.; et al. Brassinosteroid homeostasis is critical for the functionality of the Medicago truncatula pulvinus. Plant Physiol. 2021, 185, 1745–1763. [Google Scholar] [CrossRef]
- Pan, Y.H.; Michael, T.P.; Hudson, M.E.; Kay, S.A.; Chory, J.; Schuler, M.A. Cytochrome P450 Monooxygenases as Reporters for Circadian-Regulated Pathways. Plant Physiol. 2009, 150, 858–878. [Google Scholar] [CrossRef] [Green Version]
- Turk, E.M.; Fujioka, S.; Seto, H.; Shimada, Y.; Takatsuto, S.; Yoshida, S.; Denzel, M.A.; Torres, Q.I.; Neff, M.M. CYP72B1 inactivates brassinosteroid hormones: An intersection between photomorphogenesis and plant steroid signal transduction. Plant Physiol. 2003, 133, 1643–1653. [Google Scholar] [CrossRef] [Green Version]
- Neff, M.M.; Nguyen, S.M.; Malancharuvil, E.J.; Fujioka, S.; Noguchi, T.; Seto, H.; Tsubuki, M.; Honda, T.; Takatsuto, S.; Yoshida, S.; et al. BAS1: A gene regulating brassinosteroid levels and light responsiveness in Arabidopsis. Proc. Natl. Acad. Sci. USA 1999, 96, 15316–15323. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.N.; Tao, P.; Xu, F.; Wu, A.P.; Huo, W.G.; Wang, J.X. Functional Characterization of Soybean Glyma04g39610 as a Brassinosteroid Receptor Gene and Evolutionary Analysis of Soybean Brassinosteroid Receptors. Int. J. Mol. Sci. 2016, 17, 897. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| F2 Populations | No. of Total Plants | No. of WT Plants | No. of MT Plants | Expected Ratio | χ20.05 | p-Value |
|---|---|---|---|---|---|---|
| KF1 × spwp | 692 | 650 | 42 | 15:1 | 0.02 | 0.87 |
| NN1138-2 × spwp | 540 | 420 | 120 | 3:1 | 2.22 | 0.14 |
| NO. | Gene Name | Start (bp) | Stop (bp) | Annotation |
|---|---|---|---|---|
| 1 | Glyma.14g204500 | 47796602 | 47808011 | NB-ARC domain |
| 2 | Glyma.14g204600 | 47810987 | 47824929 | NB-ARC domain |
| 3 | Glyma.14g204700 | 47831269 | 47846928 | NB-ARC domain |
| 4 | Glyma.14g204800 | 47834239 | 47834920 | No annotation |
| 5 | Glyma.14g205000 | 47855678 | 47868591 | NB-ARC domain |
| 6 | Glyma.14g205100 | 47870805 | 47877043 | KIP1-like protein |
| 7 | Glyma.14g205200 | 47892074 | 47896155 | Cytochrome P450 |
| 8 | Glyma.14g205300 | 47895257 | 47907012 | NB-ARC domain |
| 9 | Glyma.14g205400 | 47909453 | 47911194 | No annotation |
| 10 | Glyma.14g205500 | 47911849 | 47919011 | Domain found in IF2B/IF5 |
| 11 | Glyma.14g205600 | 47929529 | 47931437 | AP2 domain |
| 12 | Glyma.14g205700 | 47936205 | 47940755 | Ubiquitin-conjugating enzyme |
| 13 | Glyma.14g205801 | 47939508 | 47939654 | No annotation |
| 14 | Glyma.14g206000 | 47946254 | 47952120 | Protein kinase domain |
| 15 | Glyma.14g206100 | 47972022 | 47975414 | BTB/POZ domain/NPH3 family |
| NO. | Gene Name | Start (bp) | Stop (bp) | Annotation |
|---|---|---|---|---|
| 1 | Glyma.11g228900 | 37308090 | 37313815 | Cytochrome P450 |
| 2 | Glyma.11g229000 | 37315051 | 37320857 | Nuclear pore component |
| 3 | Glyma.11g229100 | 37328504 | 37331716 | Cholesterol-capturing domain |
| 4 | Glyma.11g229300 | 37343716 | 37352599 | ABC1 family |
| 5 | Glyma.11g229400 | 37352888 | 37356662 | Sodium/hydrogen exchanger family |
| 6 | Glyma.11g229500 | 37376823 | 37382293 | GDSL-like lipase/acylhydrolase |
| 7 | Glyma.11g229600 | 37385109 | 37391331 | GINS complex protein |
| 8 | Glyma.11g229700 | 37394752 | 37395822 | Protein of unknown function (DUF1313) |
| 9 | Glyma.11g229800 | 37396251 | 37399628 | Cyclophilin type peptidyl-prolyl cis-trans isomerase/CLD |
| 10 | Glyma.11g229900 | 37404283 | 37408842 | Phosphatidylinositol-specific phospholipase C, X domain |
| 11 | Glyma.11g230000 | 37410777 | 37417244 | Phosphatidylinositol-specific phospholipase C, X domain |
| 12 | Glyma.11g230100 | 37419547 | 37424310 | Phosphatidylinositol-specific phospholipase C, X domain |
| 13 | Glyma.11g230200 | 37425247 | 37433034 | NF-X1-type zinc finger |
| 14 | Glyma.11g230300 | 37433772 | 37435676 | Protein kinase domain |
| 15 | Glyma.11g230400 | 37443779 | 37446784 | Protein kinase domain |
| 16 | Glyma.11g230500 | 37461012 | 37462619 | Protein kinase domain |
| 17 | Glyma.11g230550 | 37464702 | 37464827 | Protein kinase domain |
| 18 | Glyma.11g230600 | 37468983 | 37471869 | Protein kinase domain |
| Genes | Physical Position in Chromosome 11 (bp) a | Mutation b | Type | Conversion of AA |
|---|---|---|---|---|
| Glyma.11g229000 | 37318335 | C→T | Synonymous | / |
| Glyma.11g229400 | 37354075 | A→G | Nonsynonymous | Glu→Gly |
| Glyma.11g230000 | 37411111 | G→C | Nonsynonymous | Gly→Ala |
| 37411209 | G→C | Nonsynonymous | Val→Leu | |
| Glyma.11g230200 | 37430249 | G→C | Nonsynonymous | Gly→Ala |
| 37431363 | A→T | Nonsynonymous | Gln→His | |
| Glyma.11g230300 | 37433861 | G→C | Nonsynonymous | Val→Leu |
| 37433873 | T→C | Nonsynonymous | Tyr→His | |
| 37435019 | G→C | Nonsynonymous | Ala→Pro | |
| Glyma.11g230600 | 37469232 | T→C | Nonsynonymous | Val→Aal |
| 37469248 | T→C | Synonymous | / | |
| 37470305 | A→C | Nonsynonymous | Thr→Pro |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, K.; Xu, M.; Xu, Z.; Sharmin, R.A.; Zhang, M.; Zhao, T. Combining Fine Mapping, Whole-Genome Re-Sequencing, and RNA-Seq Unravels Candidate Genes for a Soybean Mutant with Short Petioles and Weakened Pulvini. Genes 2022, 13, 185. https://doi.org/10.3390/genes13020185
Kong K, Xu M, Xu Z, Sharmin RA, Zhang M, Zhao T. Combining Fine Mapping, Whole-Genome Re-Sequencing, and RNA-Seq Unravels Candidate Genes for a Soybean Mutant with Short Petioles and Weakened Pulvini. Genes. 2022; 13(2):185. https://doi.org/10.3390/genes13020185
Chicago/Turabian StyleKong, Keke, Mengge Xu, Zhiyong Xu, Ripa Akter Sharmin, Mengchen Zhang, and Tuanjie Zhao. 2022. "Combining Fine Mapping, Whole-Genome Re-Sequencing, and RNA-Seq Unravels Candidate Genes for a Soybean Mutant with Short Petioles and Weakened Pulvini" Genes 13, no. 2: 185. https://doi.org/10.3390/genes13020185