
Identification of HIV Rapid Mutations Using Differences in Nucleotide Distribution over Time



Abstract
: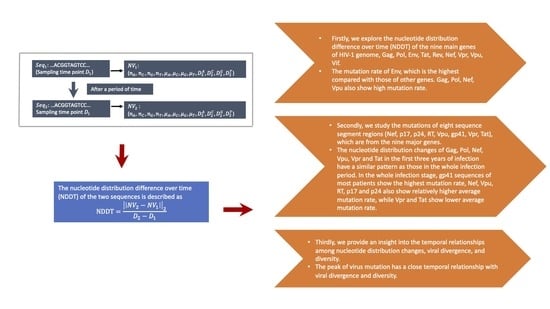
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. The Traditional 12-Dimensional Natural Vector
- .
- .
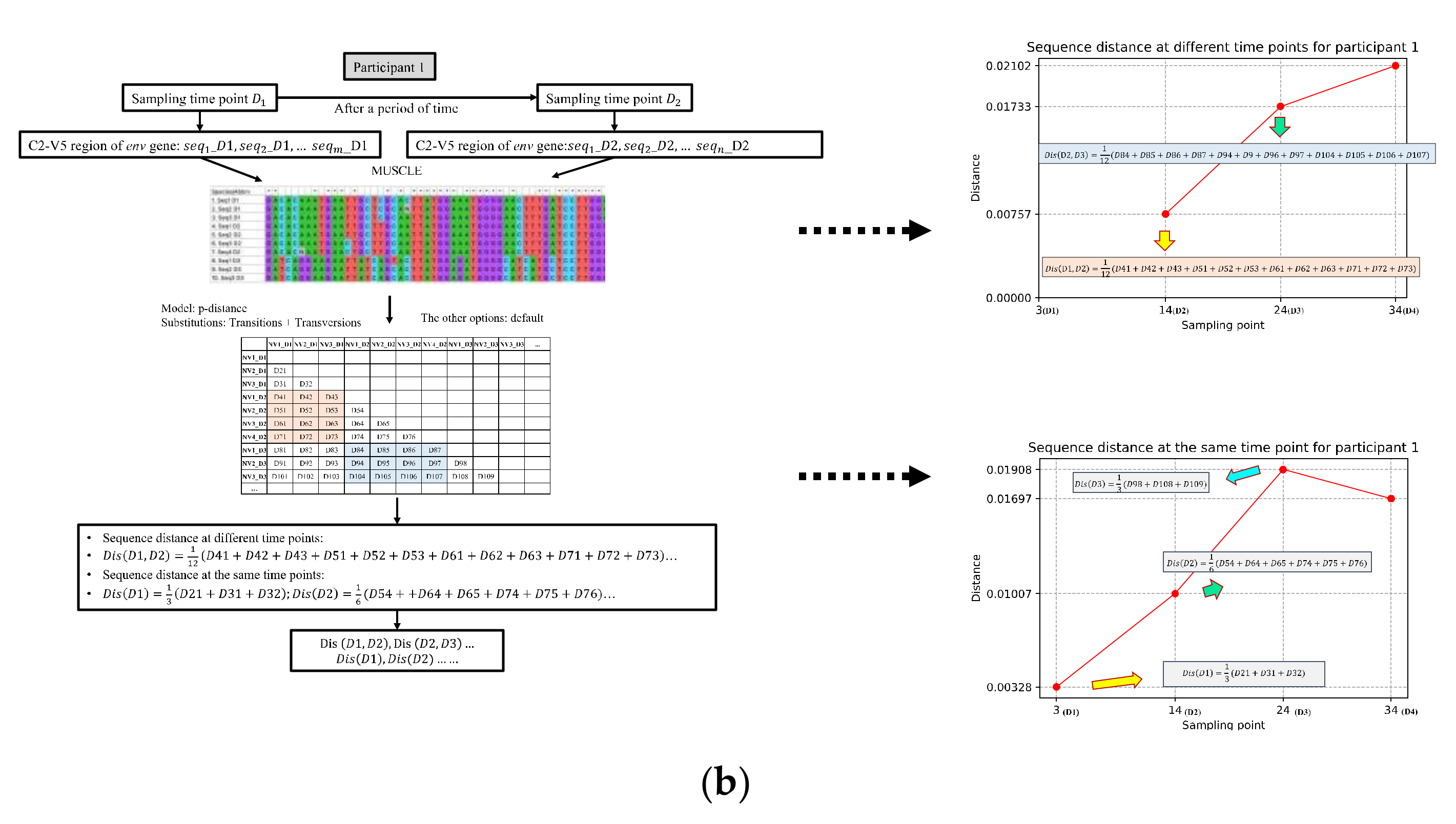
2.2. Measure the Changes of Nucleotide Distribution over Time Based on Natural Vector
2.3. Validation Dataset
3. Results
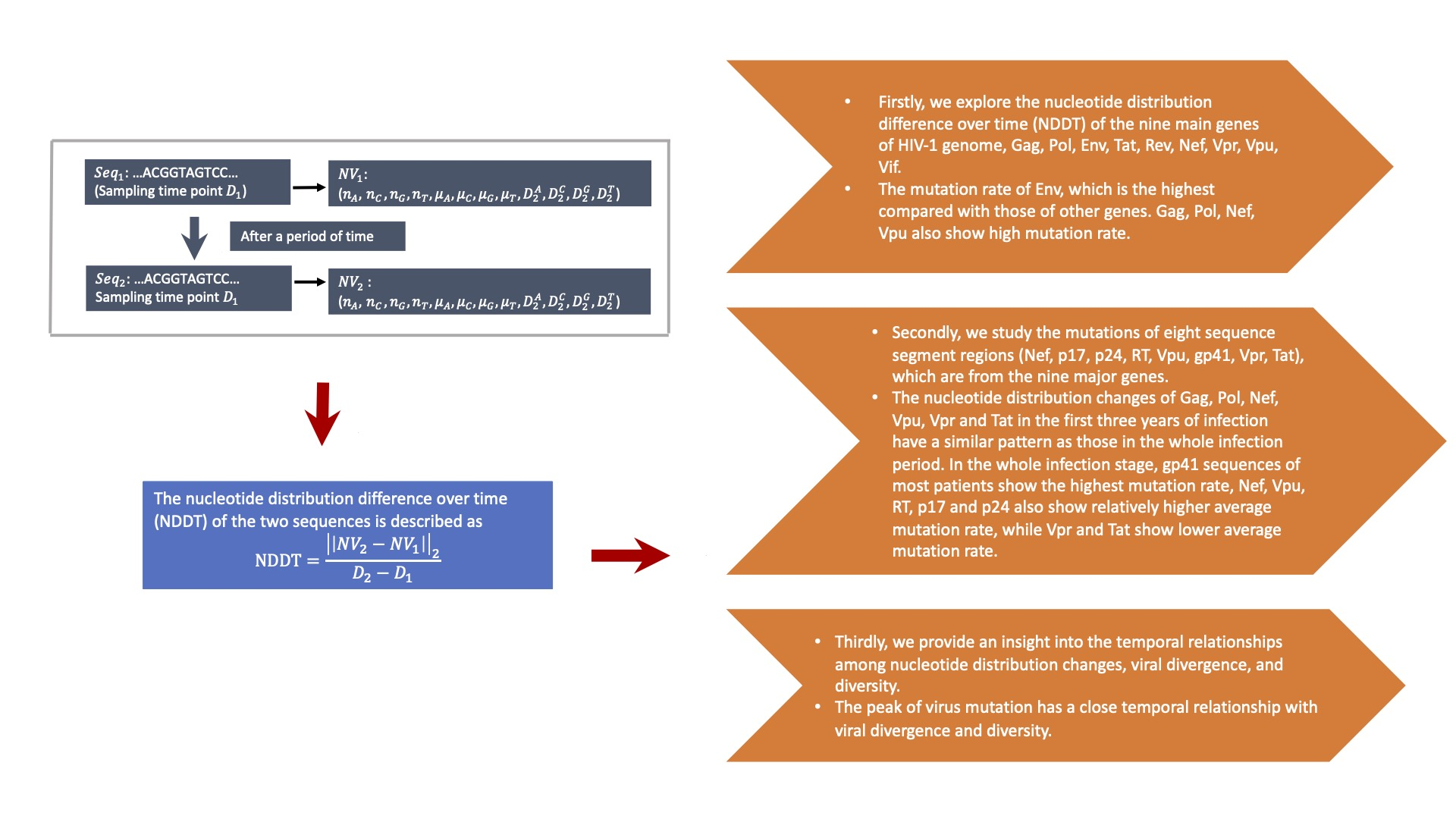
3.1. NDDT Comparison of the Nine Major Genes in HIV-1 Genome
3.2. NDDT Study of Eight Sequence Segment Regions
3.3. The Temporal Relationships among Nucleotide Distribution Changes, Viral Divergence, and Diversity
3.4. Running Time Comparison of Alignment Method and Our Alignment-Free Method
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weiss, R. How Does HIV Cause AIDS? Science 1993, 260, 1273–1279. Available online: http://www.jstor.org/stable/2881758 (accessed on 31 July 2021). [CrossRef]
- Robertson, D.L. Recombination in aids viruses. J. Mol. Evol. 1995, 40, 249–259. [Google Scholar] [CrossRef]
- Douek, D.C.; Roederer, M.; Koup, R.A. Emerging concepts in the immunopathogenesis of AIDS. Annu. Rev. Med. 2009, 60, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, P.B.; McKeague, I.W.; Eisen, G.; Mullins, C.; Guéye-Ndiaye, A.; Mboup, S.; Kanki, P.J. Comparison of HIV-1 and HIV-2 infectivity from a prospective cohort study in Senegal. Stat. Med. 2003, 22, 573–593. [Google Scholar] [CrossRef]
- UNAIDS; WHO. AIDS Epidemic Update. Available online: https://data.unaids.org/pub/epislides/2007/2007_epiupdate_en.pdf (accessed on 8 June 2021).
- Shankarappa, R.; Margolick, J.B.; Gange, S.J.; Rodrigo, A.G.; Upchurch, D.; Farzadegan, H.; Gupta, P.; Rinaldo, C.R.; Learn, G.H.; He, X.; et al. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol. 1999, 73, 10489–10502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, M.; Benková, K.; Selinger, P.; Dogoši, M.; Luňáčková, I.K.; Koutníková, H.; Laštíková, J.; Roubíčková, A.; Špůrková, Z.; Laclová, L.; et al. Opportunistic infections in HIV-infected patients differ strongly in frequencies and spectra between patients with low CD4 cell counts examined postmortem and compensated patients examined antemortem irrespective of the HAART Era. PLoS ONE 2016, 11, e0162704. [Google Scholar] [CrossRef]
- Cunningham, A.L.; Donaghy, H.; Harman, A.N.; Kim, M.; Turville, S. Manipulation of dendritic cell function by viruses. Curr. Opin. Microbiol. 2010, 13, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Kwong, P.D.; Wyatt, R.; Robinson, J.; Sweet, R.W.; Sodroski, J.; Hendrickson, W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doitsh, G.; Galloway, N.L.K.; Geng, X.; Yang, Z.; Monroe, K.M.; Zepeda, O.; Hunt, P.W.; Hatano, H.; Sowinski, S.; Muñoz-Arias, I.; et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2014, 505, 509–514. [Google Scholar] [CrossRef] [Green Version]
- WHO. HIV/AIDS. Available online: https://www.who.int/en/news-room/fact-sheets/detail/hiv-aids (accessed on 8 June 2021).
- CDC. About HIV. Available online: https://www.cdc.gov/hiv/basics/whatishiv.html (accessed on 8 June 2021).
- Kumar, V.; Abbas, A.K.; Aster, J.C. Robbins Basic Pathology; Elsevier: Amsterdam, The Netherlands, 2012; p. 147. [Google Scholar]
- Garg, H.; Mohl, J.; Joshi, A. HIV-1 induced bystander apoptosis. Viruses 2012, 4, 3020–3043. [Google Scholar] [CrossRef]
- Reeves, J.D.; Doms, R.W. Human immunodeficiency virus type 2. J. Gen. Virol. 2002, 83, 1253–1265. [Google Scholar] [CrossRef] [Green Version]
- Notredame, C. Recent Evolutions of multiple sequence alignment algorithms. PLoS Comput. Biol. 2007, 3, e123. [Google Scholar] [CrossRef] [Green Version]
- Chatzou, M.; Magis, C.; Chang, J.-M.; Kemena, C.; Bussotti, G.; Erb, I.; Notredame, C. Multiple sequence alignment modeling: Methods and applications. Brief. Bioinform. 2016, 17, 1009–1023. [Google Scholar] [CrossRef] [Green Version]
- Yin, C.; Chen, Y.; Yau, S.S.T. A measure of DNA sequence similarity by Fourier Transform with applications on hierarchical clustering. J. Theor. Biol. 2014, 359, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Zhu, Z.; Yin, C.; He, R.L.; Yau, S.S.-T. A new method to cluster genomes based on cumulative Fourier power spectrum. Gene 2018, 673, 239–250. [Google Scholar] [CrossRef]
- Pei, S.; Dong, R.; He, R.L.; Yau, S.S.-T. Large-scale genome comparison based on cumulative Fourier power and phase spectra: Central moment and covariance vector. Comput. Struct. Biotechnol. J. 2019, 17, 982–994. [Google Scholar] [CrossRef]
- Wen, J.; Chan, R.H.; Yau, S.-C.; He, R.L.; Yau, S.S. K-mer natural vector and its application to the phylogenetic analysis of genetic sequences. Gene 2014, 546, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Dong, R.; Pei, S.; Yin, C.; Yau, S.S.-T.A. A new method based on coding sequence density to cluster bacteria. J. Comput. Biol. 2020, 27, 1688–1698. [Google Scholar] [CrossRef]
- Yu, C.; Hernandez, T.; Zheng, H.; Yau, S.-C.; Huang, H.-H.; He, R.L.; Yang, J.; Yau, S.S.-T. Real time classification of viruses in 12 dimensions. PLoS ONE 2013, 8, e64328. [Google Scholar] [CrossRef] [PubMed]
- Zielezinski, A.; Girgis, H.Z.; Bernard, G.; Leimeister, C.-A.; Tang, K.; Dencker, T.; Lau, A.K.; Röhling, S.; Choi, J.J.; Waterman, M.S.; et al. Benchmarking of alignment-free sequence comparison methods. Genome Biol. 2019, 20, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Tian, K.; Yin, C.; He, R.L.; Yau, S.S.-T. Virus classification in 60-dimensional protein space. Mol. Phylogenet. Evol. 2016, 99, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Pei, S.; He, L.; Yin, C.; He, R.L.; Yau, S.S.-T. Geometric construction of viral genome space and its applications. Comput. Struct. Biotechnol. J. 2021, 19, 4226–4234. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Tian, K.; Yau, S.S.T. A new efficient method for analyzing fungi species using correlations between nucleotides. BMC Evol. Biol. 2018, 18, 200. [Google Scholar] [CrossRef] [PubMed]
- Zanini, F.; Brodin, J.; Thebo, L.; Lanz, C.; Bratt, G.; Albert, J.; Neher, R.A. Population genomics of intrapatient HIV-1 evolution. eLife 2015, 4, e11282. [Google Scholar] [CrossRef]
- Liu, Y.; McNevin, J.; Cao, J.; Zhao, H.; Genowati, I.; Wong, K.; McLaughlin, S.; McSweyn, M.D.; Diem, K.; Stevens, C.E.; et al. Selection on the Human Immunodeficiency Virus Type 1 Proteome Following Primary Infection. J. Virol. 2006, 80, 9519–9529. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; McNevin, J.; Zhao, H.; Tebit, D.M.; Troyer, R.M.; McSweyn, M.; Ghosh, A.K.; Shriner, D.; Arts, E.J.; McElrath, M.J.; et al. Evolution of Human Immunodeficiency Virus Type 1 Cytotoxic T-Lymphocyte Epitopes: Fitness-Balanced Escape. J. Virol. 2007, 81, 12179–12188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; McNevin, J.P.; Holte, S.; McElrath, M.J.; Mullins, J.I. Dynamics of Viral Evolution and CTL Responses in HIV-1 Infection. PLoS ONE 2011, 6, e15639. [Google Scholar] [CrossRef]
- Leitner, T.; Kumar, S.; Albert, J. Tempo and mode of nucleotide substitutions in gag and Env gene fragments in human immunodeficiency virus type 1 populations with a known transmission history. J. Virol. 1997, 71, 4761–4770. [Google Scholar] [CrossRef] [Green Version]
- Levy, J. Pathogenesis of human immunodeficiency virus infection. Microbiol. Rev. 1993, 57, 183–289. [Google Scholar] [CrossRef] [PubMed]
- Kaslow, R.A.; Ostrow, D.G.; Detels, R.; Phair, J.P.; Polk, B.F.; Rinaldo, C.R., Jr. The multicenter aids cohort study: Rationale, organization, and selected characteristics of the participants. Am. J. Epidemiol. 2017, 185, 1148–1156. [Google Scholar] [CrossRef]
- Yin, C. Genotyping coronavirus SARS-CoV-2: Methods and implications. Genomics 2020, 112, 3588–3596. [Google Scholar] [CrossRef]
- Chen, J.; Gao, K.; Wang, R.; Wei, G. Prediction and mitigation of mutation threats to COVID-19 vaccines and antibody therapies. Chem. Sci. 2021, 12, 6929–6948. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stecher, G.; Tamura, K.; Kumar, S. Molecular evolutionary genetics analysis (MEGA) for macOS. Mol. Biol. Evol. 2020, 37, 1237–1239. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, N.; Yang, J.; Yau, S.S.-T. Identification of HIV Rapid Mutations Using Differences in Nucleotide Distribution over Time. Genes 2022, 13, 170. https://doi.org/10.3390/genes13020170
Sun N, Yang J, Yau SS-T. Identification of HIV Rapid Mutations Using Differences in Nucleotide Distribution over Time. Genes. 2022; 13(2):170. https://doi.org/10.3390/genes13020170
Chicago/Turabian StyleSun, Nan, Jie Yang, and Stephen S.-T. Yau. 2022. "Identification of HIV Rapid Mutations Using Differences in Nucleotide Distribution over Time" Genes 13, no. 2: 170. https://doi.org/10.3390/genes13020170