
Clonal Elimination of the Pathogenic Allele as Diagnostic Pitfall in SAMD9L-Associated Neuropathy
 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
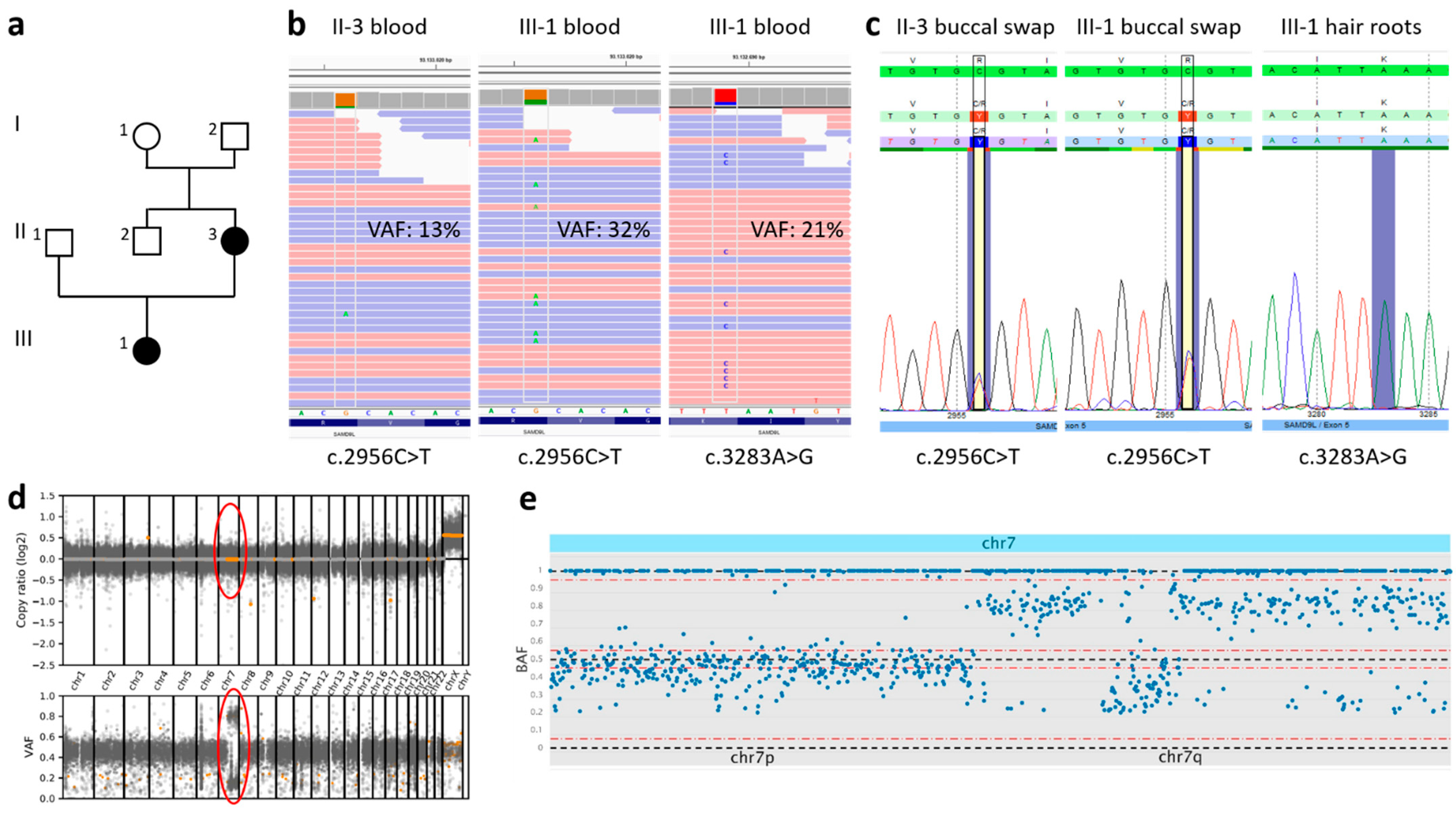
2.1. Case Presentation
2.2. Genetic Investigation
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schwartz, J.R.; Tamara, L.; Lamprecht, T.; Walsh, M.; Wang, S.; Bryant, V.; Song, G.; Wu, G.; Easton, J.; Kesserwan, C.; et al. The genomic landscape of pediatric myelodysplastic syndromes. Nat. Commun. 2017, 8, 1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastor, V.B.; Sahoo, S.S.; Boklan, J.; Schwabe, G.C.; Saribeyoglu, E.; Strahm, B.; Lebrecht, D.; Voss, M.; Bryceson, Y.T.; Erlacher, M.; et al. Constitutional SAMD9L mutations cause familial myelodysplastic syndrome and transient monosomy 7. Haematologica 2017, 103, 427–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahoo, S.S.; Pastor, V.B.; Goodings, C.; Voss, R.K.; Kozyra, E.J.; Szvetnik, A.; Noellke, P.; Dworzak, M.; Starý, J.; Locatelli, F.; et al. Clinical evolution, genetic landscape and trajectories of clonal hematopoiesis in SAMD9/SAMD9L syndromes. Nat. Med. 2021, 27, 1806–1817. [Google Scholar] [CrossRef] [PubMed]
- De Jesus, A.A.; Hou, Y.; Brooks, S.; Malle, L.; Biancotto, A.; Huang, Y.; Calvo, K.R.; Marrero, B.; Moir, S.; Oler, A.J.; et al. Distinct interferon signatures and cytokine patterns define additional systemic autoinflammatory diseases. J. Clin. Investig. 2020, 130, 1669–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.-H.; Below, J.; Shimamura, A.; Keel, S.B.; Matsushita, M.; Wolff, J.; Sul, Y.; Bonkowski, E.; Castella, M.; Taniguchi, T.; et al. Ataxia-Pancytopenia Syndrome Is Caused by Missense Mutations in SAMD9L. Am. J. Hum. Genet. 2016, 98, 1146–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorcenco, S.; Komulainen-Ebrahim, J.; Nordborg, K.; Suo-Palosaari, M.; Andréasson, S.; Krüger, J.; Nilsson, C.; Kjellström, U.; Rahikkala, E.; Turkiewicz, D.; et al. Ataxia-pancytopenia syndrome with SAMD9L mutations. Neurol. Genet. 2017, 3, e183. [Google Scholar] [CrossRef] [Green Version]
- Tesi, B.; Davidsson, J.; Voss, M.; Rahikkala, E.; Holmes, T.D.; Chiang, S.C.C.; Komulainen-Ebrahim, J.; Gorcenco, S.; Nilsson, A.R.; Ripperger, T.; et al. Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood 2017, 129, 2266–2279. [Google Scholar] [CrossRef] [Green Version]
- Cheah, J.J.; Brown, A.L.; Schreiber, A.W.; Feng, J.; Babic, M.; Moore, S.; Young, C.-C.; Fine, M.; Phillips, K.; Guandalini, M.; et al. A novel germline SAMD9L mutation in a family with ataxia-pancytopenia syndrome and pediatric acute lymphoblastic leukemia. Haematologica 2019, 104, e318–e321. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, D.; Bogdanova-Mihaylova, P.; Costello, D.J.; Sweeney, B.J.; McNamara, B.; Walsh, R.A.; Murphy, S.M. Ataxia pancytopenia syndrome due to SAMD9L mutation presenting as demyelinating neuropathy. J. Peripher. Nerv. Syst. 2020, 25, 433–437. [Google Scholar] [CrossRef]
- King-Robson, J.; Marshall, J.; Smith, F.; Willoughby, L.; Mansour, S.; Sztriha, L. Ataxia-Pancytopenia Syndrome due to a de Novo SAMD9L Mutation. Neurol. Genet. 2021, 7, e580. [Google Scholar] [CrossRef]
- Sainio, M.T.; Aaltio, J.; Hyttinen, V.; Kortelainen, M.; Ojanen, S.; Paetau, A.; Tienari, P.; Ylikallio, E.; Auranen, M.; Tyynismaa, H. Effectiveness of clinical exome sequencing in adult patients with difficult-to-diagnose neurological disorders. Acta Neurol. Scand. 2021, 145, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Corral-Juan, M.; Casquero, P.; Giraldo-Restrepo, N.; Laurie, S.; Martinez-Piñeiro, A.; Mateo-Montero, R.C.; Ispierto, L.; Vilas, D.; Tolosa, E.; Volpini, V.; et al. New spinocerebellar ataxia subtype caused by SAMD9L mutation triggering mitochondrial dysregulation (SCA49). Brain Commun. 2022, 4, fcac030. [Google Scholar] [CrossRef] [PubMed]
- Allenspach, E.J.; Soveg, F.; Finn, L.S.; So, L.; Gorman, J.A.; Rosen, A.B.; Skoda-Smith, S.; Wheeler, M.M.; Barrow, K.A.; Rich, L.M.; et al. Germline SAMD9L truncation variants trigger global translational repression. J. Exp. Med. 2021, 218, e20201195. [Google Scholar] [CrossRef] [PubMed]
- Davidsson, J.; Puschmann, A.; Tedgård, U.; Bryder, D.; Nilsson, L.; Cammenga, J. SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia 2018, 32, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Garrison, E.; Marth, G. Haplotype-Based Variant Detection from Short-Read Sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Guo, Y.; Ding, X.; Shen, Y.; Lyon, G.J.; Wang, K. SeqMule: Automated pipeline for analysis of human exome/genome sequencing data. Sci. Rep. 2015, 5, srep14283. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; O Pollard, M.; Whitwham, A.; Keane, T.; A McCarthy, S.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- van Riet, J.; Krol, N.M.; Atmodimedjo, P.N.; Brosens, E.; van Ijcken, W.F.; Jansen, M.P.; Martens, J.W.; Looijenga, L.H.; Jenster, G.; Dubbink, H.J.; et al. SNPitty. J. Mol. Diagn. 2018, 20, 166–176. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.C.; Bryant, V.; Lamprecht, T.; Ma, J.; Walsh, M.; Schwartz, J.; Alzamora, M.D.P.; Mullighan, C.G.; Loh, M.L.; Ribeiro, R.; et al. Germline SAMD9 and SAMD9L mutations are associated with extensive genetic evolution and diverse hematologic outcomes. J. Clin. Investig. 2018, 3, e121086. [Google Scholar] [CrossRef]
- Collin, M. I am SAMD9L: 7q regulator I am. Blood 2017, 129, 2210–2212. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eggermann, K.; Meyer, R.; Begemann, M.; Dey, D.; Bültmann, E.; Kurth, I.; Korenke, G.C.; Knopp, C. Clonal Elimination of the Pathogenic Allele as Diagnostic Pitfall in SAMD9L-Associated Neuropathy. Genes 2022, 13, 2356. https://doi.org/10.3390/genes13122356
Eggermann K, Meyer R, Begemann M, Dey D, Bültmann E, Kurth I, Korenke GC, Knopp C. Clonal Elimination of the Pathogenic Allele as Diagnostic Pitfall in SAMD9L-Associated Neuropathy. Genes. 2022; 13(12):2356. https://doi.org/10.3390/genes13122356
Chicago/Turabian StyleEggermann, K., R. Meyer, M. Begemann, D. Dey, E. Bültmann, I. Kurth, G. C. Korenke, and C. Knopp. 2022. "Clonal Elimination of the Pathogenic Allele as Diagnostic Pitfall in SAMD9L-Associated Neuropathy" Genes 13, no. 12: 2356. https://doi.org/10.3390/genes13122356