
A Comprehensive Sequencing Analysis of Testis-Born miRNAs in Immature and Mature Indigenous Wandong Cattle (Bos taurus)
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Experimental Animals, Sample Collection and Histological Assessment
2.3. Small-RNA Library Construction for Illumina
2.4. Illumina Sequencing and Bioinformatics Analysis
2.5. Known and Novel miRNAs Prediction
2.6. Differentially Expressed miRNA Analysis
2.7. GO and KEGG Enrichment Analysis
2.8. Integrated Analysis among miRNAs–mRNAs and DE genes
2.9. Real Time Quantitative PCR
2.10. Statistical Analysis
3. Results
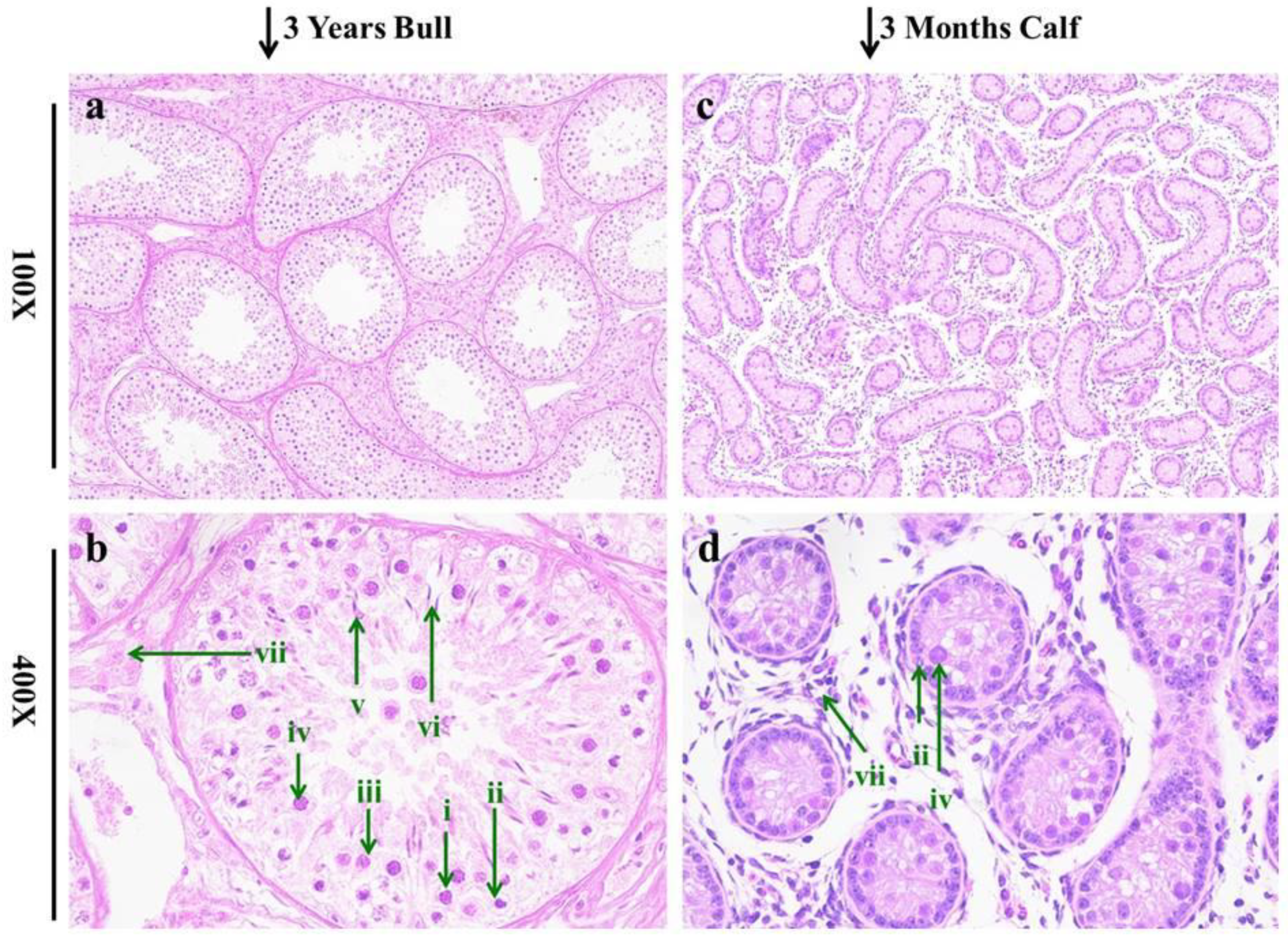
3.1. Histomorphology of Testis
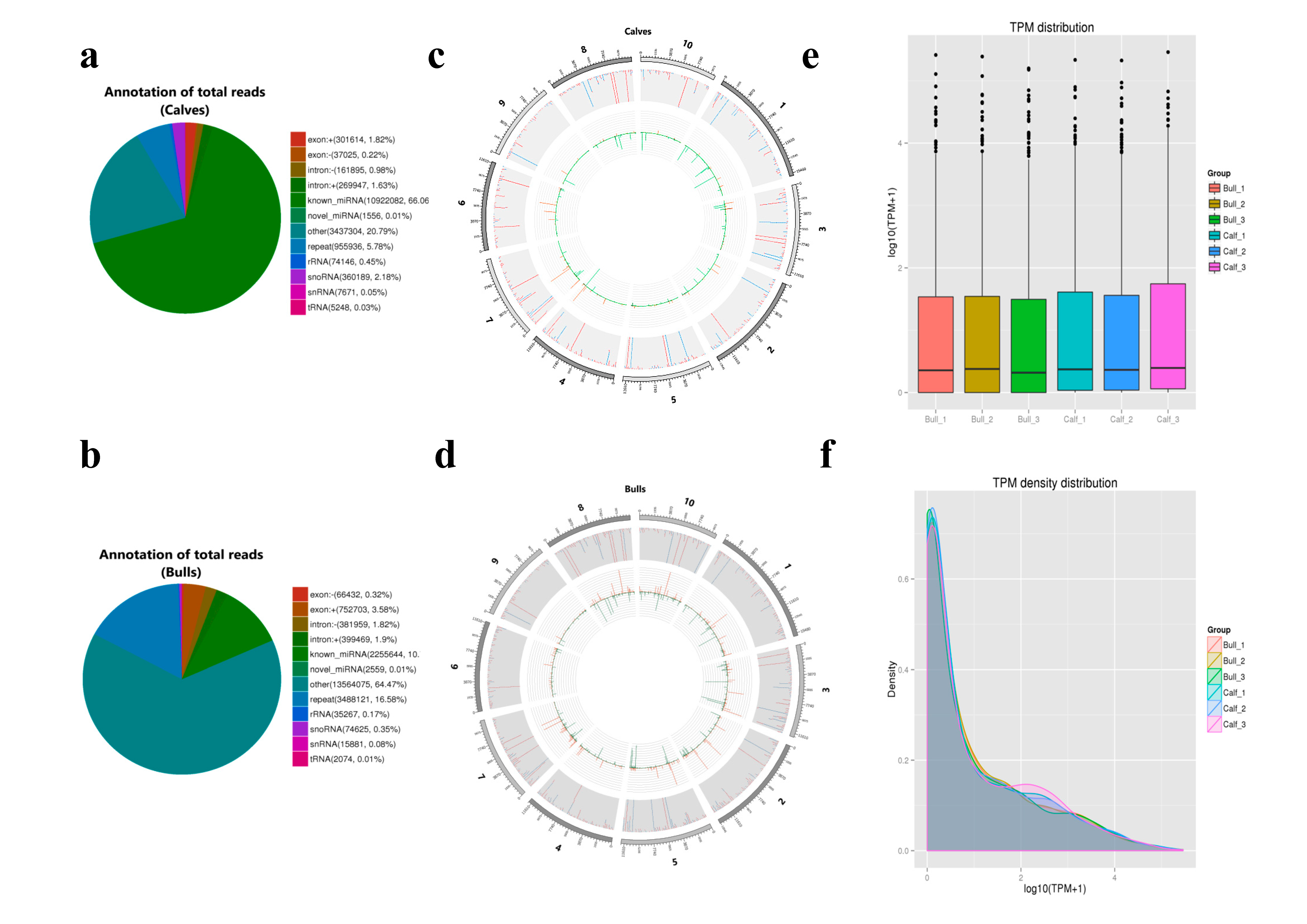
3.2. Transcriptomics Summary of Sequencing Data
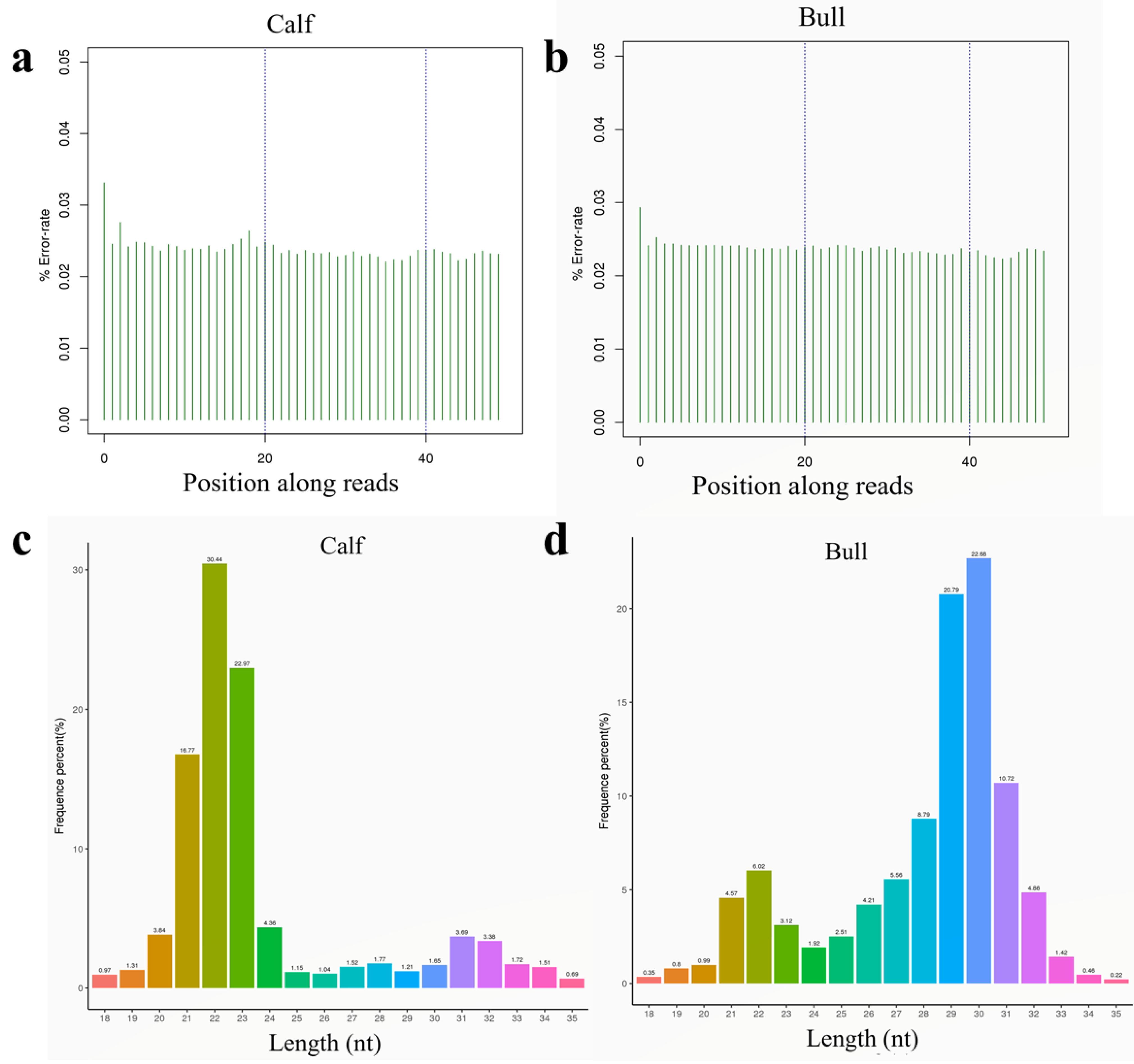
3.3. Sequencing Data Quality
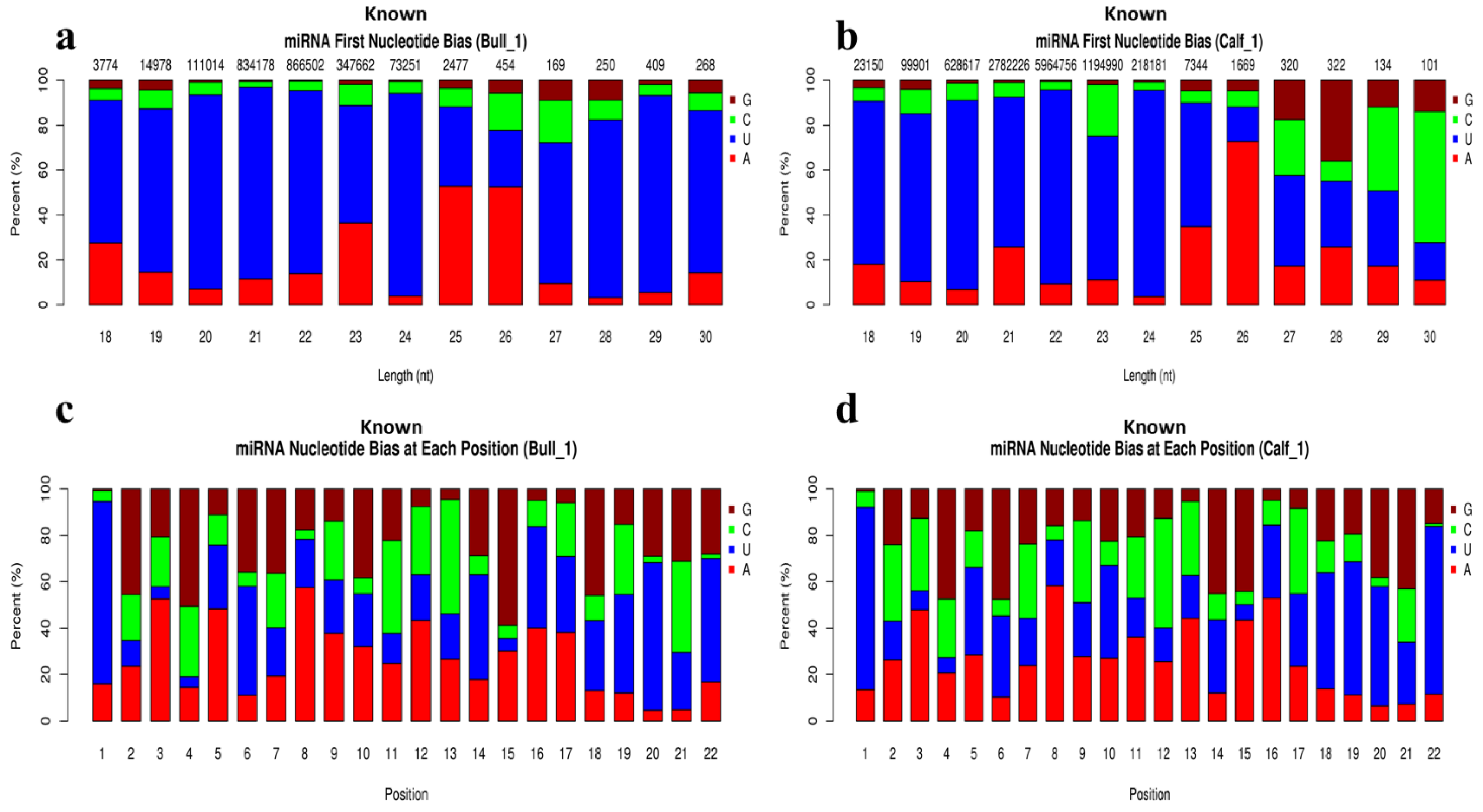
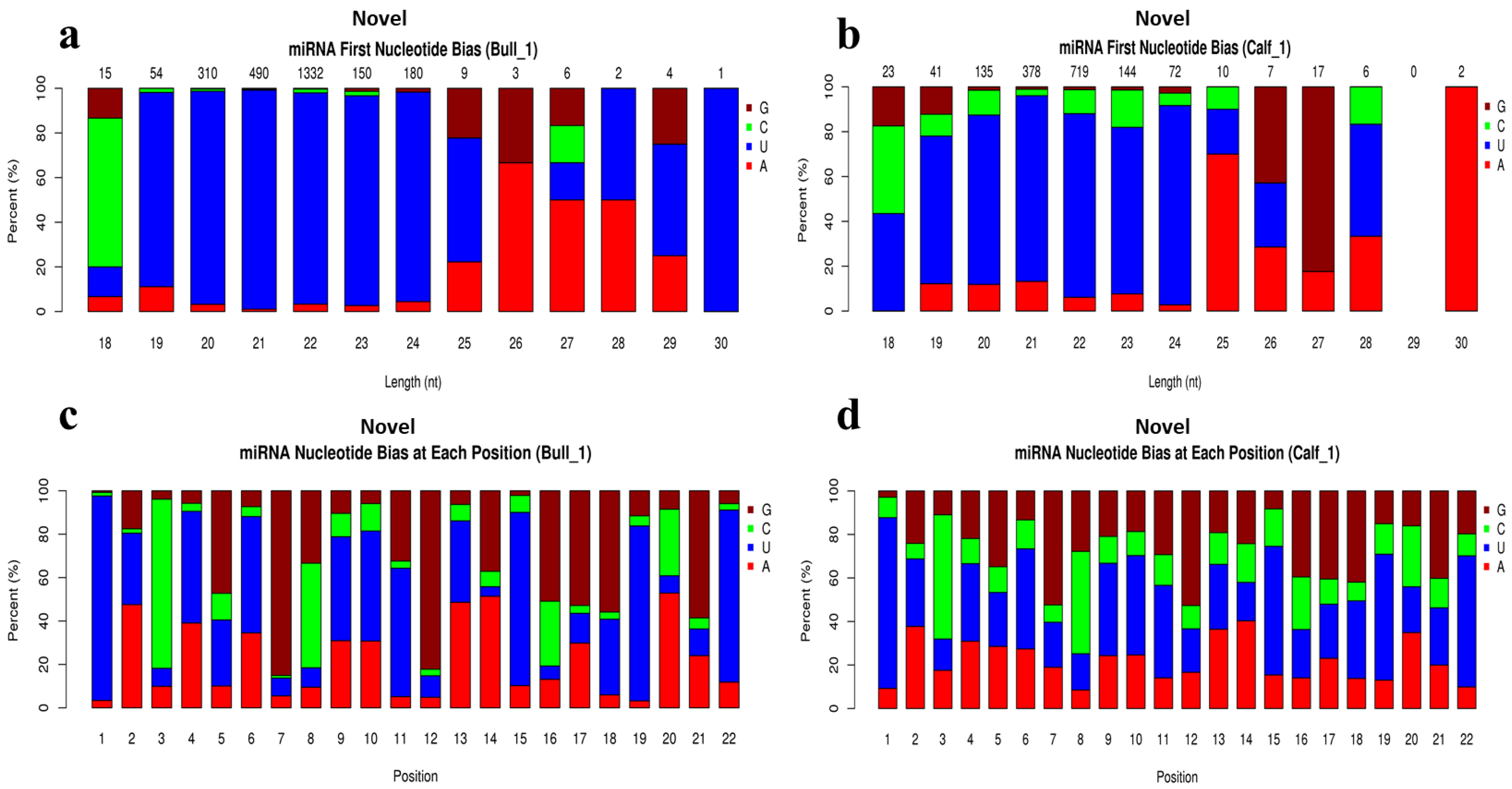
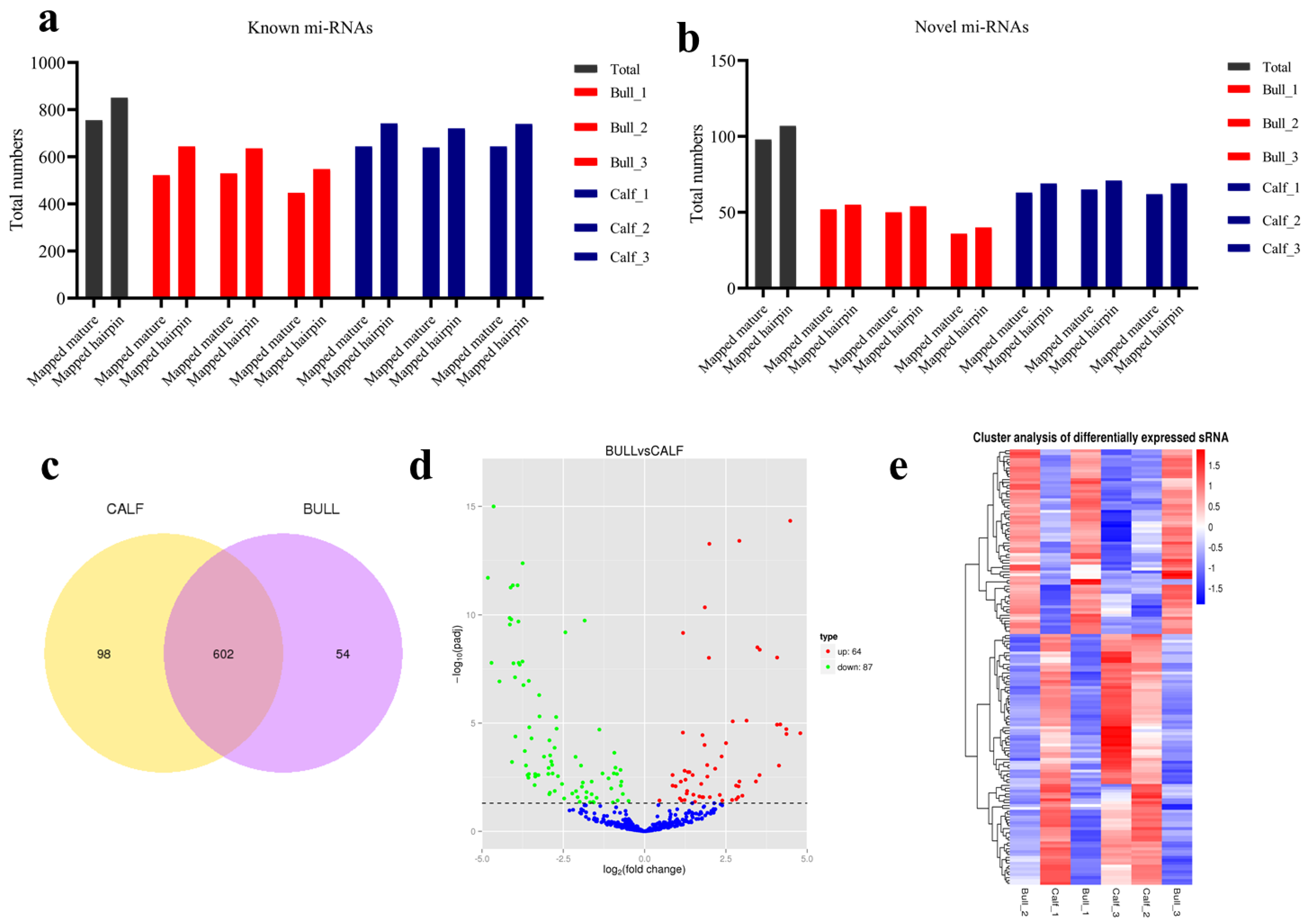
3.4. Analysis of Known and Novel miRNAs
3.5. Characterization of miRNAs
3.6. miRNAs Expression Profiling of Bovine Testis in Different Developmental Stages
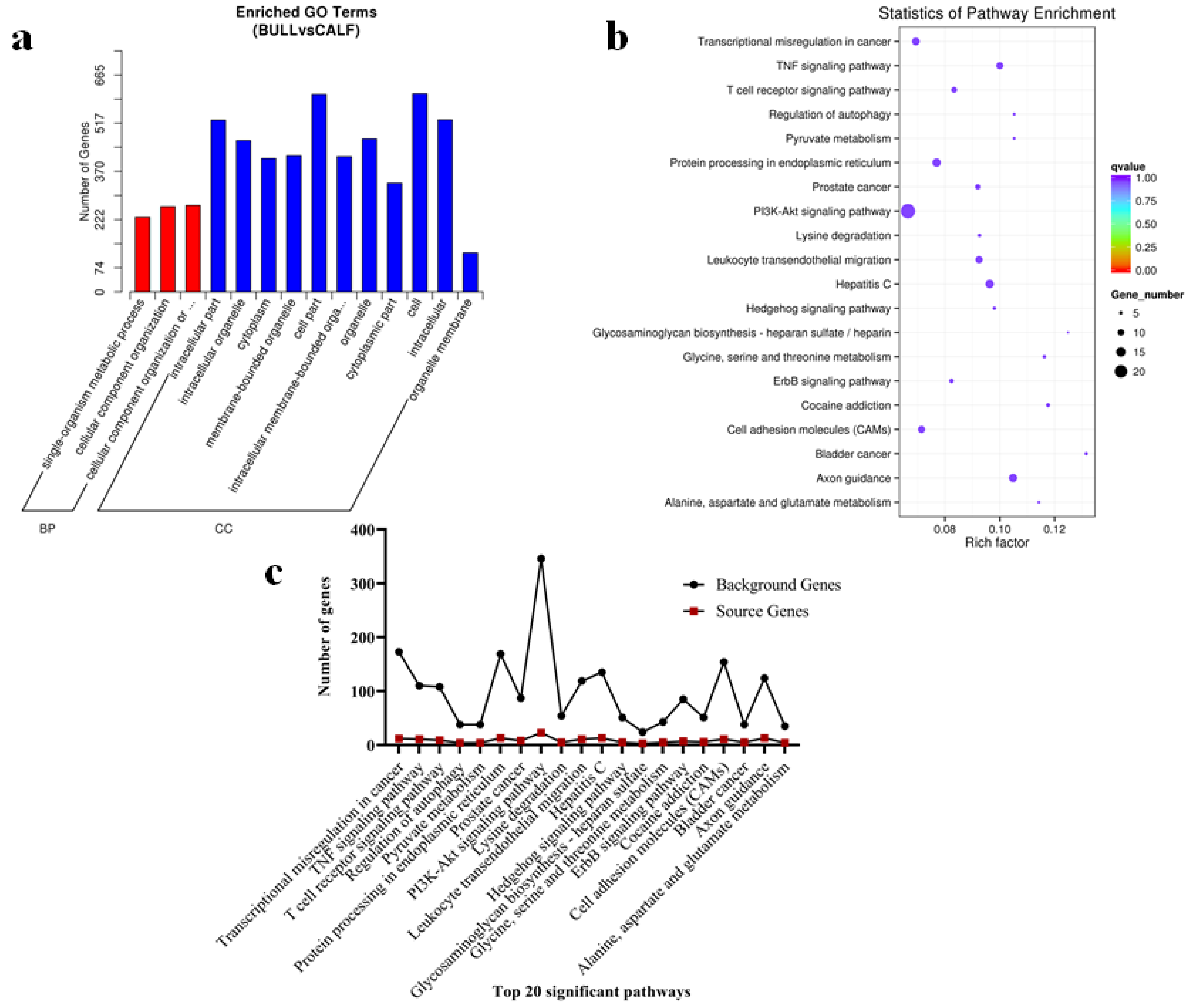
3.7. The miRNAs Targets, GO and KEGG Pathways Analysis
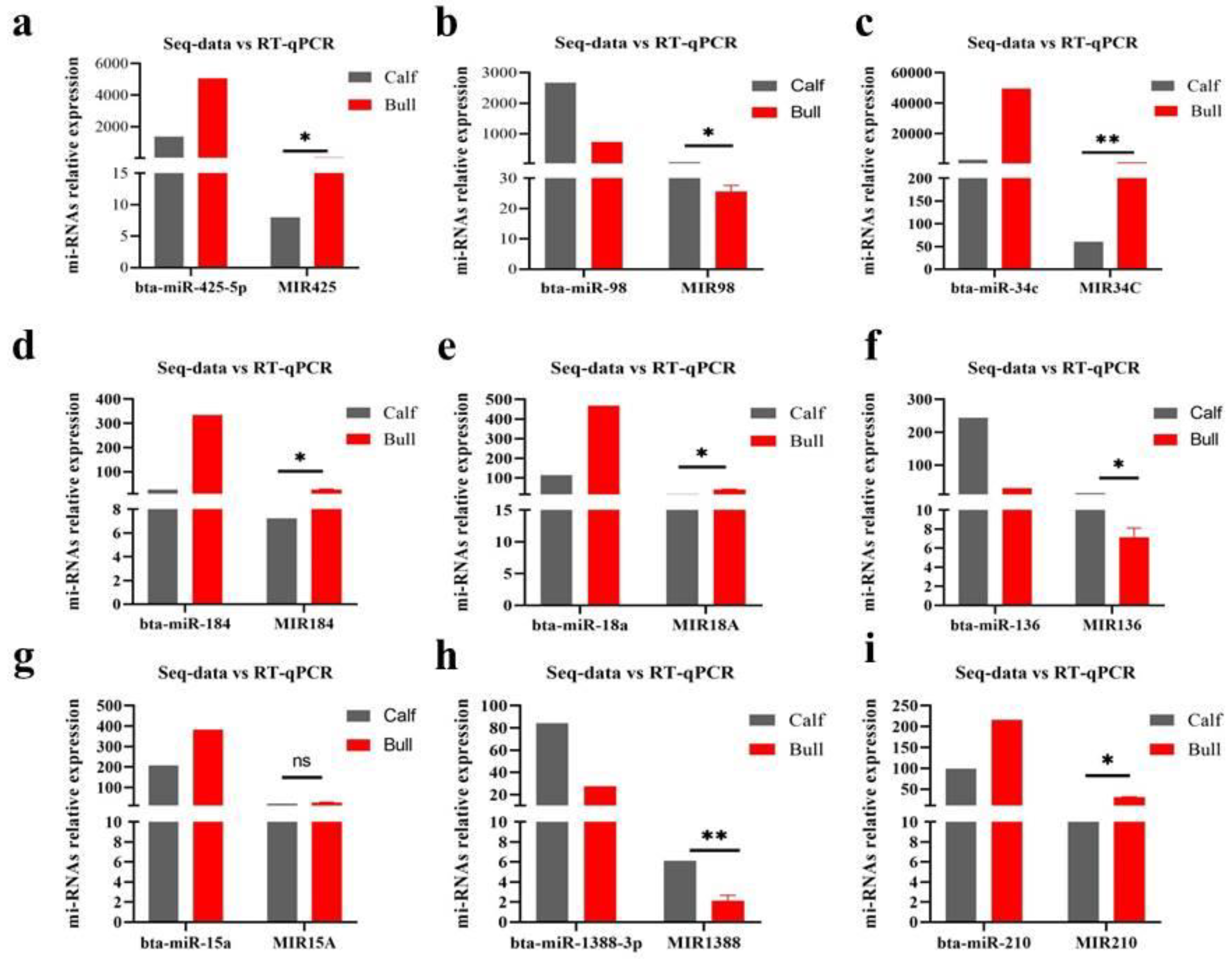
3.8. Validation of DE miRNAs Seq-Results by RT-qPCR
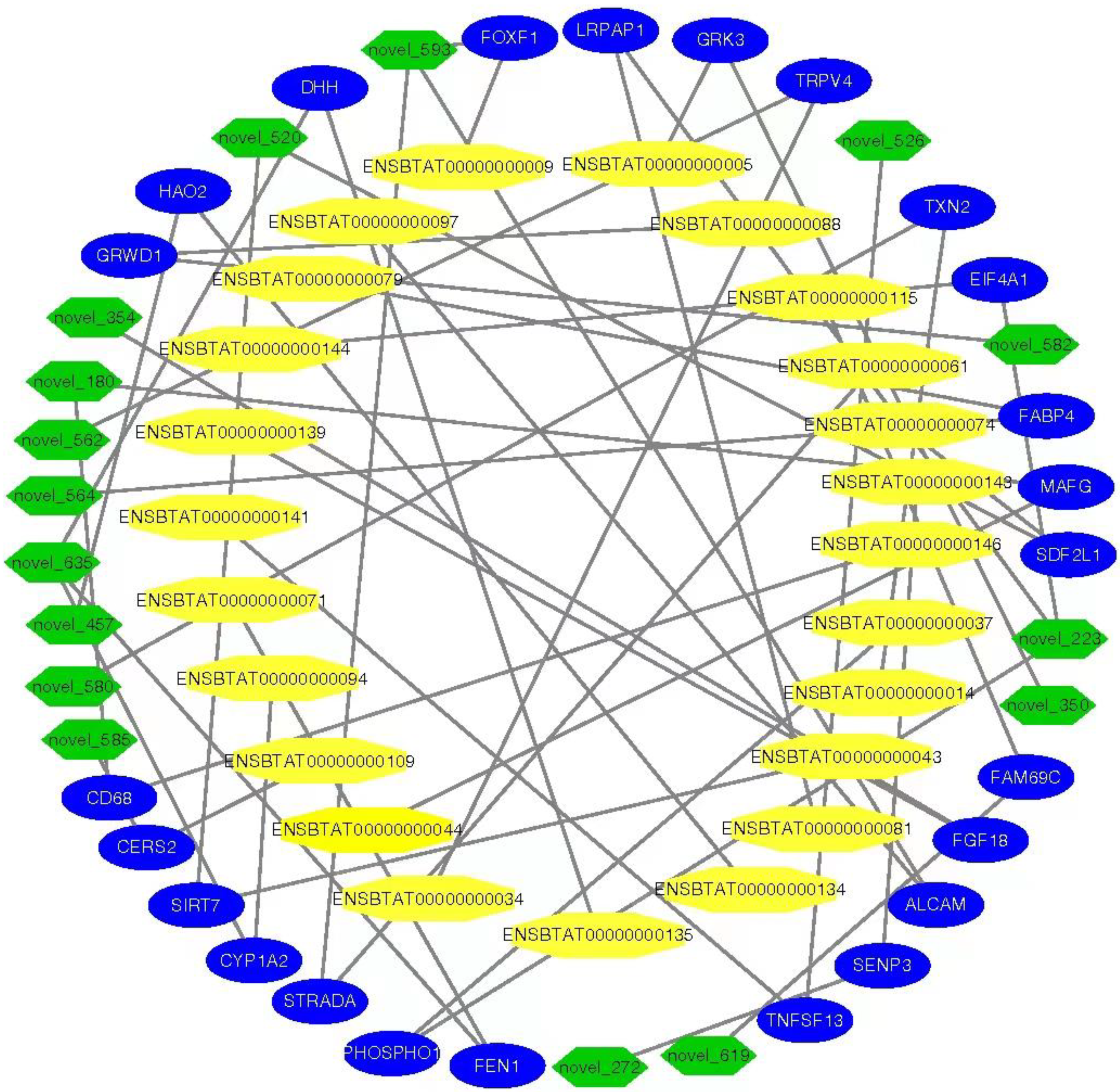
3.9. MiRNAs–mRNAs and Link Genes Interaction Analysis
3.10. Functional Differentially Expressed Genes (DEGs)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Svingen, T.; Koopman, P. Building the mammalian testis: Origins, differentiation, and assembly of the component cell populations. Genes Dev. 2013, 27, 2409–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Khan, I.M.; Yin, H.; Zhou, X.; Rizwan, M.; Zhuang, J.; Zhang, Y. Integrated Analysis of Long Non-Coding RNA and mRNA Expression Profiles in Testes of Calves and Sexually Mature Wandong Bulls (Bos taurus). Animals 2021, 11, 2006. [Google Scholar] [CrossRef] [PubMed]
- Ansari-Lari, M.; Kafi, M.; Sokhtanlo, M.; Ahmadi, H.N. Reproductive performance of Holstein dairy cows in Iran. Trop. Anim. Health Prod. 2010, 42, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Peñagaricano, F. Unravelling the genomic architecture of bull fertility in Holstein cattle. BMC Genet. 2016, 17, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Li, L.; Jiang, H.; Ma, J.E.; Li, J.; Chen, J. Identification and differential expression of microRNAs in testis and ovary of Amur sturgeon (Acipenser schrenckii). Gene 2018, 658, 36–46. [Google Scholar] [CrossRef]
- Brennecke, J.; Hipfner, D.R.; Stark, A.; Russell, R.B.; Cohen, S.M. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell 2003, 113, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Lei, J.; Ding, L.; Wen, Y.; Ju, H.; Zhang, X. MicroRNA: Function, detection, and bioanalysis. Chem. Rev. 2013, 113, 6207–6233. [Google Scholar] [CrossRef]
- Guil, S.; Esteller, M. RNA–RNA interactions in gene regulation: The coding and noncoding players. Trends Biochem. Sci. 2015, 40, 248–256. [Google Scholar] [CrossRef]
- Bushati, N.; Cohen, S.M. microRNA functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 175–205. [Google Scholar] [CrossRef]
- Curry, E.; Safranski, T.J.; Pratt, S.L. Differential expression of porcine sperm microRNAs and their association with sperm morphology and motility. Theriogenology 2011, 76, 1532–1539. [Google Scholar] [CrossRef]
- Hayashi, K.; Chuva de Sousa Lopes, S.M.; Kaneda, M.; Tang, F.; Hajkova, P.; Lao, K.; O’Carroll, D.; Das, P.P.; Tarakhovsky, A.; Miska, E.A. MicroRNA biogenesis is required for mouse primordial germ cell development and spermatogenesis. PLoS ONE 2008, 3, e1738. [Google Scholar] [CrossRef] [Green Version]
- Hong, X.; Luense, L.J.; McGinnis, L.K.; Nothnick, W.B.; Christenson, L.K. Dicer1 is essential for female fertility and normal development of the female reproductive system. Endocrinology 2008, 149, 6207–6212. [Google Scholar] [CrossRef] [Green Version]
- Suh, N.; Baehner, L.; Moltzahn, F.; Melton, C.; Shenoy, A.; Chen, J.; Blelloch, R. MicroRNA function is globally suppressed in mouse oocytes and early embryos. Curr. Biol. 2010, 20, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Niu, Z.; Li, Q.; Pang, R.T.; Chiu, P.C.; Yeung, W.S.B. Micro RNA and embryo implantation. Am. J. Reprod. Immunol. 2016, 75, 263–271. [Google Scholar] [CrossRef]
- Vidigal, J.A.; Ventura, A. Embryonic stem cell miRNAs and their roles in development and disease. In Proceedings of the Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2012; pp. 428–436. [Google Scholar]
- Cook, M.S.; Blelloch, R. Small RNAs in germline development. Curr. Top. Dev. Biol. 2013, 102, 159–205. [Google Scholar]
- Otsuka, M.; Zheng, M.; Hayashi, M.; Lee, J.-D.; Yoshino, O.; Lin, S.; Han, J. Impaired microRNA processing causes corpus luteum insufficiency and infertility in mice. J. Clin. Investig. 2008, 118, 1944–1954. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Liu, Q.; Zhang, W.; Li, J.; Li, Z.; Tang, Z.; Li, Y.; Han, C.; Hall, S.H.; Zhang, Y. Comparative profiling of genes and miRNAs expressed in the newborn, young adult, and aged human epididymides. Acta Biochim. Biophys. Sin. 2010, 42, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Liu, Y.; Wang, T.; Guan, J.; Luo, Z.; Chen, H.; Wang, X.; Chen, L.; Ma, J.; Mu, Z. Repertoire of porcine microRNAs in adult ovary and testis by deep sequencing. Int. J. Biol. Sci. 2011, 7, 1045. [Google Scholar] [CrossRef] [Green Version]
- Luo, L.; Ye, L.; Liu, G.; Shao, G.; Zheng, R.; Ren, Z.; Zuo, B.; Xu, D.; Lei, M.; Jiang, S. Microarray-based approach identifies differentially expressed microRNAs in porcine sexually immature and mature testes. PLoS ONE 2010, 5, e11744. [Google Scholar] [CrossRef]
- Torley, K.J.; da Silveira, J.C.; Smith, P.; Anthony, R.V.; Veeramachaneni, D.; Winger, Q.A.; Bouma, G.J. Expression of miRNAs in ovine fetal gonads: Potential role in gonadal differentiation. Reprod. Biol. Endocrinol. 2011, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Eggers, S.; Ohnesorg, T.; Sinclair, A. Genetic regulation of mammalian gonad development. Nat. Rev. Endocrinol. 2014, 10, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Rakoczy, J.; Fernandez-Valverde, S.L.; Glazov, E.A.; Wainwright, E.N.; Sato, T.; Takada, S.; Combes, A.N.; Korbie, D.J.; Miller, D.; Grimmond, S.M. MicroRNAs-140-5p/140-3p modulate Leydig cell numbers in the developing mouse testis. Biol. Reprod. 2013, 88, 143. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhong, J.; Chai, Z.; Zhu, J.; Xin, J. Comparative expression profile of micro RNA s and pi RNA s in three ruminant species testes using next-generation sequencing. Reprod. Domest. Anim. 2018, 53, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wu, S.; Zhao, W.; Mipam, T.; Liu, J.; Liu, W.; Yi, C.; Yu, S.; Cai, X. Differentially expressed microRNAs between cattleyak and yak testis. Sci. Rep. 2018, 8, 592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; ali Shah, M.; Mipam, T.; Wu, S.; Yi, C.; Luo, H.; Yuan, M.; Chai, Z.; Zhao, W.; Cai, X. Bovid microRNAs involved in the process of spermatogonia differentiation into spermatocytes. Int. J. Biol. Sci. 2020, 16, 239. [Google Scholar] [CrossRef] [Green Version]
- Lunstra, D.-D.; Echternkamp, S. Puberty in beef bulls: Acrosome morphology and semen quality in bulls of different breeds. J. Anim. Sci. 1982, 55, 638–648. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.H.; Jacobson, K.A.; Rose, J.; Zeller, R. Hematoxylin and eosin staining of tissue and cell sections. Cold Spring Harb. Protoc. 2008, 2008, pdb.prot4986. [Google Scholar] [CrossRef]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Wen, M.; Shen, Y.; Shi, S.; Tang, T. miREvo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinform. 2012, 13, 140. [Google Scholar] [CrossRef]
- Zhou, L.; Chen, J.; Li, Z.; Li, X.; Hu, X.; Huang, Y.; Zhao, X.; Liang, C.; Wang, Y.; Sun, L. Integrated profiling of microRNAs and mRNAs: microRNAs located on Xq27. 3 associate with clear cell renal cell carcinoma. PLoS ONE 2010, 5, e15224. [Google Scholar] [CrossRef]
- Enright, A.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D. MicroRNA targets in Drosophila. Genome Biol. 2003, 4, R1. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Zheng, X.; Yang, J.; Imamichi, T.; Stephens, R.; Lempicki, R.A. Extracting biological meaning from large gene lists with DAVID. Curr. Protoc. Bioinform. 2009, 27, 13.11. [Google Scholar] [CrossRef]
- Wrobel, K.-H.; Schimmel, M. Morphology of the bovine Sertoli cell during the spermatogenetic cycle. Cell Tissue Res. 1989, 257, 93–103. [Google Scholar] [CrossRef]
- Vasileva, A.; Tiedau, D.; Firooznia, A.; Müller-Reichert, T.; Jessberger, R. Tdrd6 is required for spermiogenesis, chromatoid body architecture, and regulation of miRNA expression. Curr. Biol. 2009, 19, 630–639. [Google Scholar] [CrossRef] [Green Version]
- Buchold, G.M.; Coarfa, C.; Kim, J.; Milosavljevic, A.; Gunaratne, P.H.; Matzuk, M.M. Analysis of microRNA expression in the prepubertal testis. PLoS ONE 2010, 5, e15317. [Google Scholar] [CrossRef] [Green Version]
- Gong, W.; Pan, L.; Lin, Q.; Zhou, Y.; Xin, C.; Yu, X.; Cui, P.; Hu, S.; Yu, J. Transcriptome profiling of the developing postnatal mouse testis using next-generation sequencing. Sci. China Life Sci. 2013, 56, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Luo, Y.; Liu, M.; Huang, J.; Xu, D. Histological and transcriptome analyses of testes from Duroc and Meishan boars. Sci. Rep. 2016, 6, 20758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.; Wang, J.; Wang, K.; Luo, Y.; Tang, Q.; Liu, X.; Fang, M. Integrated analysis of miRNA-mRNA network reveals different regulatory patterns in the endometrium of Meishan and Duroc sows during mid-late gestation. Animals 2020, 10, 420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.; Chen, Q.; Gao, Y.; Li, J.; Li, W.; Dang, R.; Lei, C. Comparative transcriptomics analysis of testicular miRNA from cryptorchid and normal horses. Animals 2020, 10, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; He, X.; Zhao, Y.; Bai, D.; Du, M.; Song, L.; Liu, Z.; Yin, Z.; Manglai, D. Transcriptome profiling of developing testes and spermatogenesis in the Mongolian horse. BMC Genet. 2020, 21, 46. [Google Scholar] [CrossRef]
- Bai, M.; Sun, L.; Jia, C.; Li, J.; Han, Y.; Liu, H.; Chen, Y.; Jiang, H. Integrated analysis of miRNA and mRNA expression profiles reveals functional miRNA-targets in development testes of small tail han sheep. G3 Genes Genomes Genet. 2019, 9, 523–533. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, J.; Fang, C.; Shi, L.; Tan, J.; Xiong, Y.; Fan, B.; Li, C. Genome-wide differential expression of genes and small RNAs in testis of two different porcine breeds and at two different ages. Sci. Rep. 2016, 6, 26852. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Ju, Z.; Li, Q.; Hou, Q.; Wang, C.; Li, J.; Li, R.; Wang, L.; Sun, T.; Hang, S. Solexa sequencing of novel and differentially expressed microRNAs in testicular and ovarian tissues in Holstein cattle. Int. J. Biol. Sci. 2011, 7, 1016. [Google Scholar] [CrossRef]
- Noveski, P.; Popovska-Jankovic, K.; Kubelka-Sabit, K.; Filipovski, V.; Lazarevski, S.; Plaseski, T.; Plaseska-Karanfilska, D. Micro RNA expression profiles in testicular biopsies of patients with impaired spermatogenesis. Andrology 2016, 4, 1020–1027. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Guo, L.; Wang, J.; Li, M.; Appiah, M.O.; Liu, H.; Zhao, J.; Yang, L.; Lu, W. Photoperiodic effect on the testicular transcriptome in broiler roosters. J. Anim. Physiol. Anim. Nutr. 2020, 104, 918–927. [Google Scholar] [CrossRef]
- Lu, Q.; Gore, M.; Zhang, Q.; Camenisch, T.; Boast, S.; Casagranda, F.; Lai, C.; Skinner, M.K.; Klein, R.; Matsushima, G.K. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature 1999, 398, 723–728. [Google Scholar] [CrossRef]
- Nowacka-Woszuk, J.; Szczerbal, I.; Stachowiak, M.; Dzimira, S.; Nizanski, W.; Biezynski, J.; Nowak, T.; Gogulski, M.; Switonski, M. Screening for structural variants of four candidate genes in dogs with disorders of sex development revealed the first case of a large deletion in NR5A1. Anim. Reprod. Sci. 2020, 223, 106632. [Google Scholar] [CrossRef]
- Gungor-Ordueri, N.E.; Mruk, D.D.; Wan, H.-t.; Wong, E.W.; Celik-Ozenci, C.; Lie, P.P.; Cheng, C.Y. New insights into FAK function and regulation during spermatogenesis. Histol. Histopathol. 2014, 29, 977. [Google Scholar]
- Guan, Y.; Liang, G.; Hawken, P.A.; Malecki, I.A.; Cozens, G.; Vercoe, P.E.; Martin, G.B.; Guan, L.L. Roles of small RNAs in the effects of nutrition on apoptosis and spermatogenesis in the adult testis. Sci. Rep. 2015, 5, 10372. [Google Scholar] [CrossRef] [Green Version]
- Tscherner, A.; Gilchrist, G.; Smith, N.; Blondin, P.; Gillis, D.; LaMarre, J. MicroRNA-34 family expression in bovine gametes and preimplantation embryos. Reprod. Biol. Endocrinol. 2014, 12, 85. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Yu, M.; Liu, C.; Zhu, H.; He, X.; Peng, S.; Hua, J. miR-34c works downstream of p53 leading to dairy goat male germline stem-cell (mGSC s) apoptosis. Cell Prolif. 2013, 46, 223–231. [Google Scholar] [CrossRef]
- Wu, J.; Bao, J.; Wang, L.; Hu, Y.; Xu, C. MicroRNA-184 downregulates nuclear receptor corepressor 2 in mouse spermatogenesis. BMC Dev. Biol. 2011, 11, 64. [Google Scholar] [CrossRef] [Green Version]
- Xie, R.; Lin, X.; Du, T.; Xu, K.; Shen, H.; Wei, F.; Hao, W.; Lin, T.; Lin, X.; Qin, Y. Targeted disruption of miR-17-92 impairs mouse spermatogenesis by activating mTOR signaling pathway. Medicine 2016, 95, e2713. [Google Scholar] [CrossRef]
- Yang, F.; Guan, J.; Li, R.; Li, X.; Niu, J.; Shang, R.; Qi, J.; Wang, X. miR-1388 regulates the expression of nectin2l in Paralichthys olivaceus. Comp. Biochem. Physiol. Part D Genom. Proteom. 2018, 28, 9–16. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Micro-RNA | Gen-Bank. No | Forward Primer | Reverse-Primer | Tm |
|---|---|---|---|---|
| bta-miR-425-5p | NR_030967.1 | GCCGAGATGACACGATCACT | Universal Fix-Seq | 55 °C |
| bta-miR-98 | NR_031360.1 | GCCGAGTGAGGTAGTAAGTT | Universal Fix-Seq | 55 °C |
| bta-miR-34c | NR_030942.1 | GCCGAG AGGCAGTGTAGTTA | Universal Fix-Seq | 55 °C |
| bta-miR-184 | NR_031179.1 | GCCGAG TGGACGGAGAACTG | Universal Fix-Seq | 55 °C |
| bta-miR-18a | NR_030892.1 | GCCGAGTAAGGTGCATCTAG | Universal Fix-Seq | 55 °C |
| bta-miR-136 | NR_030865.1 | GCCGAGACTCCATTTGTTTTG | Universal Fix-Seq | 60 °C |
| bta-miR-15a | NR_030793.1 | GCCGAGTAGCAGCACATAA | Universal Fix-Seq | 65 °C |
| bta-miR-1388-3p | NR_036335.1 | GCCGAG ATCTCAGGTTTGTUC | Universal Fix-Seq | 65 °C |
| bta-miR-210 | NR_049717.1 | GCCGAGACTGTGCGTGTGACA | Universal Fix-Seq | 65 °C |
| Sample | Reads | Bases | Error Rate | Q20 | Q30 | GC Content |
|---|---|---|---|---|---|---|
| Bull_1 | 23004713 | 1.150G | 0.01% | 98.57% | 95.35% | 48.78% |
| Bull_2 | 19243007 | 0.962G | 0.01% | 99.25% | 97.13% | 48.71% |
| Bull_3 | 21312453 | 1.066G | 0.01% | 98.48% | 95.11% | 48.32% |
| Calf_1 | 17862525 | 0.893G | 0.01% | 98.51% | 95.28% | 48.84% |
| Calf_2 | 17696661 | 0.885G | 0.01% | 99.35% | 97.56% | 48.79% |
| Calf_3 | 21796590 | 1.090G | 0.01% | 98.49% | 95.07% | 48.99% |
| Groups | Terms | DEGs No | GO_Accession | Genes ID |
|---|---|---|---|---|
| Calf vs. Bull | Sexual reproduction | 14 | 0019953 | CFAP157, SEPT6, CCDC36, MEI1, FOLR2, BBS1, TYRO3, B4GALNT1, BCL6, BSP3, JAG2, PHC2, TGFBR1, CTDNEP1 |
| Male gonad development | 4 | 0008584 | NR5A1, TGFBR1, SF1, NKX3-1 | |
| Germ cell development | 4 | 0007281 | SH2B1, CDKN1A, HYAL1, MYOG | |
| Germ cell migration | 1 | 0008354 | TGFBR1 | |
| Spermatid development | 3 | 0007286 | MEI1, BSP3, CFAP157 | |
| Fusion of sperm to egg plasma membrane | 1 | 0007342 | FOLR2 | |
| Sperm part | 3 | 0097223 | TMEM190, SEPTIN6, TEKT3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Khan, I.M.; Liu, Y.; Khan, N.M.; Ji, K.; Yin, H.; Wang, W.; Zhou, X.; Zhang, Y. A Comprehensive Sequencing Analysis of Testis-Born miRNAs in Immature and Mature Indigenous Wandong Cattle (Bos taurus). Genes 2022, 13, 2185. https://doi.org/10.3390/genes13122185
Liu H, Khan IM, Liu Y, Khan NM, Ji K, Yin H, Wang W, Zhou X, Zhang Y. A Comprehensive Sequencing Analysis of Testis-Born miRNAs in Immature and Mature Indigenous Wandong Cattle (Bos taurus). Genes. 2022; 13(12):2185. https://doi.org/10.3390/genes13122185
Chicago/Turabian StyleLiu, Hongyu, Ibrar Muhammad Khan, Yong Liu, Nazir Muhammad Khan, Kaiyuan Ji, Huiqun Yin, Wenliang Wang, Xinqi Zhou, and Yunhai Zhang. 2022. "A Comprehensive Sequencing Analysis of Testis-Born miRNAs in Immature and Mature Indigenous Wandong Cattle (Bos taurus)" Genes 13, no. 12: 2185. https://doi.org/10.3390/genes13122185