
Targeted Panel Sequencing Identifies an Intronic c.5225-3C>G Variant of the FBN1 Gene Causing Sporadic Marfan Syndrome with Annuloaortic Ectasia
 and
and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Targeted Panel Sequencing
2.2. Verification of the FBN1 c.5225-3C>G Variant
2.3. Ribonucleic Acid Isolation, Reverse Transcription PCR, and Complementary DNA Sequencing
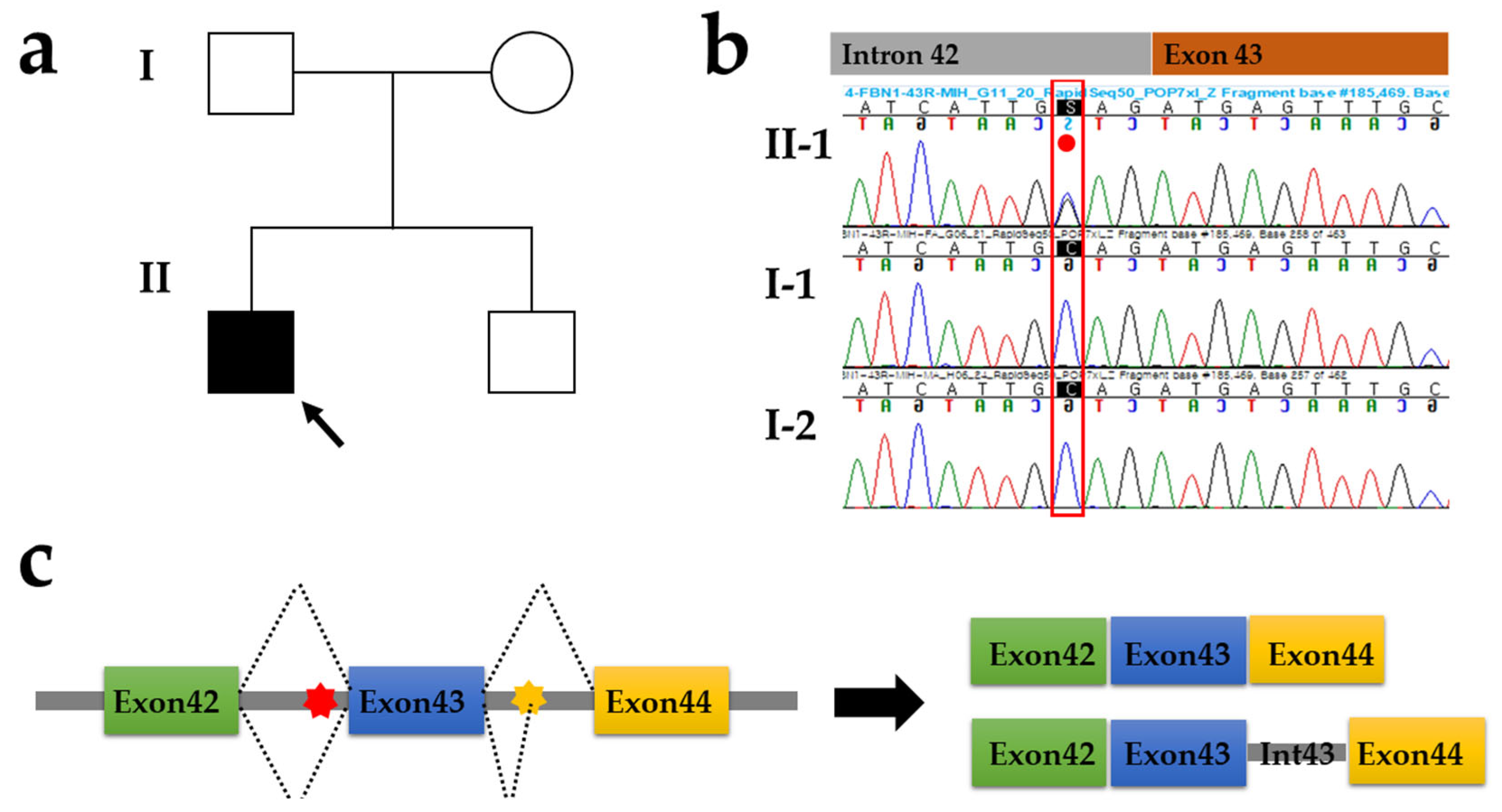
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stark, V.C.; Hensen, F.; Kutsche, K.; Kortüm, F.; Olfe, J.; Wiegand, P.; von Kodolitsch, Y.; Kozlik-Feldmann, R.; Müller, G.C.; Mir, T.S. Genotype-Phenotype Correlation in Children: The Impact of FBN1 Variants on Pediatric Marfan Care. Genes 2020, 11, 799. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, P.; Milleron, O.; Hanna, N.; Ropers, J.; Ouali, N.O.; Affoune, A.; Langeois, M.; Eliahou, L.; Arnoult, F.; Renard, P.; et al. Clinical relevance of genotype-phenotype correlations beyond vascular events in a cohort study of 1500 Marfan syndrome patients with FBN1 pathogenic variants. Genet. Med. 2021, 23, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Hernándiz, A.; Zúñiga, A.; Valera, F.; Domingo, D.; Ontoria-Oviedo, I.; Marí, J.F.; Román, J.A.; Calvo, I.; Insa, B.; Gómez, R.; et al. Genotype FBN1/phenotype relationship in a cohort of patients with Marfan syndrome. Clin. Genet. 2021, 99, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, A.; Alaerts, M.; Van Laer, L.; Loeys, B. Marfan Syndrome and Related Disorders: 25 Years of Gene Discovery. Hum. Mutat. 2016, 37, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.Y.; Seo, S.R.; Park, S.W.; Kim, D.K. The Prevalence of Marfan Syndrome in Korea. J. Korean Med. Sci. 2017, 32, 576–580. [Google Scholar] [CrossRef]
- Rybczynski, M.; Bernhardt, A.M.; Rehder, U.; Fuisting, B.; Meiss, L.; Voss, U.; Habermann, C.; Detter, C.; Robinson, P.N.; Arslan-Kirchner, M.; et al. The spectrum of syndromes and manifestations in individuals screened for suspected Marfan syndrome. Am. J. Med. Genet. A 2008, 146a, 3157–3166. [Google Scholar] [CrossRef]
- Loeys, B.; De Backer, J.; Van Acker, P.; Wettinck, K.; Pals, G.; Nuytinck, L.; Coucke, P.; De Paepe, A. Comprehensive molecular screening of the FBN1 gene favors locus homogeneity of classical Marfan syndrome. Hum. Mutat. 2004, 24, 140–146. [Google Scholar] [CrossRef]
- Landis, B.J.; Veldtman, G.R.; Ware, S.M. Genotype-phenotype correlations in Marfan syndrome. Heart 2017, 103, 1750–1752. [Google Scholar] [CrossRef] [Green Version]
- Becerra-Muñoz, V.M.; Gómez-Doblas, J.J.; Porras-Martín, C.; Such-Martínez, M.; Crespo-Leiro, M.G.; Barriales-Villa, R.; de Teresa-Galván, E.; Jiménez-Navarro, M.; Cabrera-Bueno, F. The importance of genotype-phenotype correlation in the clinical management of Marfan syndrome. Orphanet. J. Rare Dis. 2018, 13, 16. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Li, L.; Fu, Y.; Wang, X.; Sun, H.; Wang, J.; Han, L.; Wu, Z.; Liu, Y.; Zhu, J.; et al. Increased frequency of FBN1 frameshift and nonsense mutations in Marfan syndrome patients with aortic dissection. Mol. Genet. Genom. Med. 2020, 8, e1041. [Google Scholar] [CrossRef]
- Faivre, L.; Collod-Beroud, G.; Loeys, B.L.; Child, A.; Binquet, C.; Gautier, E.; Callewaert, B.; Arbustini, E.; Mayer, K.; Arslan-Kirchner, M.; et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: An international study. Am. J. Hum. Genet. 2007, 81, 454–466. [Google Scholar] [CrossRef] [Green Version]
- Faivre, L.; Collod-Beroud, G.; Callewaert, B.; Child, A.; Binquet, C.; Gautier, E.; Loeys, B.L.; Arbustini, E.; Mayer, K.; Arslan-Kirchner, M.; et al. Clinical and mutation-type analysis from an international series of 198 probands with a pathogenic FBN1 exons 24-32 mutation. Eur. J. Hum. Genet. 2009, 17, 491–501. [Google Scholar] [CrossRef]
- Wooderchak-Donahue, W.; VanSant-Webb, C.; Tvrdik, T.; Plant, P.; Lewis, T.; Stocks, J.; Raney, J.A.; Meyers, L.; Berg, A.; Rope, A.F.; et al. Clinical utility of a next generation sequencing panel assay for Marfan and Marfan-like syndromes featuring aortopathy. Am. J. Med. Genet. A 2015, 167a, 1747–1757. [Google Scholar] [CrossRef]
- Gentilini, D.; Oliveri, A.; Fazia, T.; Pini, A.; Marelli, S.; Bernardinelli, L.; Di Blasio, A.M. NGS analysis in Marfan syndrome spectrum: Combination of rare and common genetic variants to improve genotype-phenotype correlation analysis. PLoS ONE 2019, 14, e0222506. [Google Scholar] [CrossRef]
- Kayhan, G.; Ergun, M.A.; Ergun, S.G.; Kula, S.; Percin, F.E. Identification of Three Novel FBN1 Mutations and Their Phenotypic Relationship of Marfan Syndrome. Genet. Test Mol. Biomark. 2018, 22, 474–480. [Google Scholar] [CrossRef]
- Torrado, M.; Maneiro, E.; Trujillo-Quintero, J.P.; Evangelista, A.; Mikhailov, A.T.; Monserrat, L. A Novel Heterozygous Intronic Mutation in the FBN1 Gene Contributes to FBN1 RNA Missplicing Events in the Marfan Syndrome. Biomed. Res. Int. 2018, 2018, 3536495. [Google Scholar] [CrossRef] [Green Version]
- Oh, M.R.; Kim, J.S.; Beck, N.S.; Yoo, H.W.; Lee, H.J.; Kohsaka, T.; Jin, D.K. Six novel mutations of the fibrillin-1 gene in Korean patients with Marfan syndrome. Pediatr. Int. 2000, 42, 488–491. [Google Scholar] [CrossRef]
- Yoo, E.H.; Woo, H.; Ki, C.S.; Lee, H.J.; Kim, D.K.; Kang, I.S.; Park, P.; Sung, K.; Lee, C.S.; Chung, T.Y.; et al. Clinical and genetic analysis of Korean patients with Marfan syndrome: Possible ethnic differences in clinical manifestation. Clin. Genet. 2010, 77, 177–182. [Google Scholar] [CrossRef]
- Song, Y.H.; Kim, G.H.; Yoo, H.W.; Kim, J.B. Novel de novo nonsense mutation of FBN1 gene in a patient with Marfan syndrome. J. Genet. 2012, 91, 233–235. [Google Scholar] [CrossRef]
- Nam, H.K.; Nam, M.H.; Ha, K.S.; Rhie, Y.J.; Lee, K.H. A Novel Fibrillin-1 Gene Mutation Leading to Marfan Syndrome in a Korean Girl. Ann. Clin. Lab. Sci. 2017, 47, 221–225. [Google Scholar]
- Seo, G.H.; Kim, Y.M.; Kang, E.; Kim, G.H.; Seo, E.J.; Lee, B.H.; Choi, J.H.; Yoo, H.W. The phenotypic heterogeneity of patients with Marfan-related disorders and their variant spectrums. Medicine 2018, 97, e10767. [Google Scholar] [CrossRef]
- Yoon, S.H.; Kong, Y. Severe neonatal Marfan syndrome with a novel mutation in the intron of the FBN1 gene: A case report. Medicine 2021, 100, e24301. [Google Scholar] [CrossRef] [PubMed]
- Nayak, S.S.; Schneeberger, P.E.; Patil, S.J.; Arun, K.M.; Suresh, P.V.; Kiran, V.S.; Siddaiah, S.; Maiya, S.; Venkatachalagupta, S.K.; Kausthubham, N.; et al. Clinically relevant variants in a large cohort of Indian patients with Marfan syndrome and related disorders identified by next-generation sequencing. Sci. Rep. 2021, 11, 764. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e524. [Google Scholar] [CrossRef] [Green Version]
- Mannucci, L.; Luciano, S.; Salehi, L.B.; Gigante, L.; Conte, C.; Longo, G.; Ferradini, V.; Piumelli, N.; Brancati, F.; Ruvolo, G.; et al. Mutation analysis of the FBN1 gene in a cohort of patients with Marfan Syndrome: A 10-year single center experience. Clin. Chim. Acta 2020, 501, 154–164. [Google Scholar] [CrossRef]
- Bombardieri, E.; Rohrbach, M.; Greutmann, M.; Matyas, G.; Weber, R.; Radulovic, J.; Boillat, M.F.; Linka, A.; De Pasquale, G.; Bonassin, F.; et al. Marfan syndrome and related connective tissue disorders in the current era in Switzerland in 103 patients: Medical and surgical management and impact of genetic testing. Swiss Med. Wkly. 2020, 150, w20189. [Google Scholar] [CrossRef] [Green Version]
- Stengl, R.; Bors, A.; Ágg, B.; Pólos, M.; Matyas, G.; Molnár, M.J.; Fekete, B.; Csabán, D.; Andrikovics, H.; Merkely, B.; et al. Optimising the mutation screening strategy in Marfan syndrome and identifying genotypes with more severe aortic involvement. Orphanet. J. Rare Dis. 2020, 15, 290. [Google Scholar] [CrossRef]
- Overwater, E.; Marsili, L.; Baars, M.J.H.; Baas, A.F.; van de Beek, I.; Dulfer, E.; van Hagen, J.M.; Hilhorst-Hofstee, Y.; Kempers, M.; Krapels, I.P.; et al. Results of next-generation sequencing gene panel diagnostics including copy-number variation analysis in 810 patients suspected of heritable thoracic aortic disorders. Hum. Mutat. 2018, 39, 1173–1192. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhuang, X.; Chen, B.; Wen, J.; Peng, F.; Liu, X.; Wei, M. 99-Case Study of Sporadic Aortic Dissection by Whole Exome Sequencing Indicated Novel Disease-Associated Genes and Variants in Chinese Population. Biomed. Res. Int. 2020, 2020, 7857043. [Google Scholar] [CrossRef]
- Li, J.; Yang, L.; Diao, Y.; Zhou, L.; Xin, Y.; Jiang, L.; Li, R.; Wang, J.; Duan, W.; Liu, J. Genetic testing and clinical relevance of patients with thoracic aortic aneurysm and dissection in northwestern China. Mol. Genet. Genom. Med. 2021, 9, e1800. [Google Scholar] [CrossRef]
- Di, C.; Syafrizayanti; Zhang, Q.; Chen, Y.; Wang, Y.; Zhang, X.; Liu, Y.; Sun, C.; Zhang, H.; Hoheisel, J.D. Function, clinical application, and strategies of Pre-mRNA splicing in cancer. Cell Death Differ. 2019, 26, 1181–1194. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Sun, Y.; Liu, M.; Zhang, X.; Jiang, T. Functional Characterization of Argininosuccinate Lyase Gene Variants by Mini-Gene Splicing Assay. Front. Genet. 2019, 10, 436. [Google Scholar] [CrossRef]
- Wu, Y.; Sun, H.; He, Y.; Zhang, H. A novel intron mutation in FBN-1 gene identified in a pregnant woman with Marfan syndrome. Hereditas 2021, 158, 6. [Google Scholar] [CrossRef]
- Wang, W.J.; Han, P.; Zheng, J.; Hu, F.Y.; Zhu, Y.; Xie, J.S.; Guo, J.; Zhang, Z.; Dong, J.; Zheng, G.Y.; et al. Exon 47 skipping of fibrillin-1 leads preferentially to cardiovascular defects in patients with thoracic aortic aneurysms and dissections. J. Mol. Med. 2013, 91, 37–47. [Google Scholar] [CrossRef]
- Wypasek, E.; Potaczek, D.P.; Hydzik, M.; Stapor, R.; Raczkowska-Muraszko, M.; Weiss, J.; Maugeri, A.; Undas, A. Detection and a functional characterization of the novel FBN1 intronic mutation underlying Marfan syndrome: Case presentation. Clin. Chem. Lab. Med. 2018, 56, 87–91. [Google Scholar] [CrossRef]
- Fusco, C.; Morlino, S.; Micale, L.; Ferraris, A.; Grammatico, P.; Castori, M. Characterization of Two Novel Intronic Variants Affecting Splicing in FBN1-Related Disorders. Genes 2019, 10, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeyer, K.A.; Reinhardt, D.P. Engineered mutations in fibrillin-1 leading to Marfan syndrome act at the protein, cellular and organismal levels. Mutat. Res. Rev. Mutat. Res. 2015, 765, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Groth, K.A.; Gaustadnes, M.; Thorsen, K.; Østergaard, J.R.; Jensen, U.B.; Gravholt, C.H.; Andersen, N.H. Difficulties in diagnosing Marfan syndrome using current FBN1 databases. Genet. Med. 2016, 18, 98–102. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.H.; Kim, T.Y.; Kim, S.J.; Cho, Y.G.; Park, J.; Jang, W. Targeted Panel Sequencing Identifies an Intronic c.5225-3C>G Variant of the FBN1 Gene Causing Sporadic Marfan Syndrome with Annuloaortic Ectasia. Genes 2022, 13, 2108. https://doi.org/10.3390/genes13112108
Kim KH, Kim TY, Kim SJ, Cho YG, Park J, Jang W. Targeted Panel Sequencing Identifies an Intronic c.5225-3C>G Variant of the FBN1 Gene Causing Sporadic Marfan Syndrome with Annuloaortic Ectasia. Genes. 2022; 13(11):2108. https://doi.org/10.3390/genes13112108
Chicago/Turabian StyleKim, Kyung Hwa, Tae Yun Kim, Soon Jin Kim, Yong Gon Cho, Joonhong Park, and Woori Jang. 2022. "Targeted Panel Sequencing Identifies an Intronic c.5225-3C>G Variant of the FBN1 Gene Causing Sporadic Marfan Syndrome with Annuloaortic Ectasia" Genes 13, no. 11: 2108. https://doi.org/10.3390/genes13112108