
A Novel GCK Large Genomic Rearrangement in a Patient with MODY-2 Detected by Clinical Exome Sequencing

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient History
2.2. Molecular Analysis
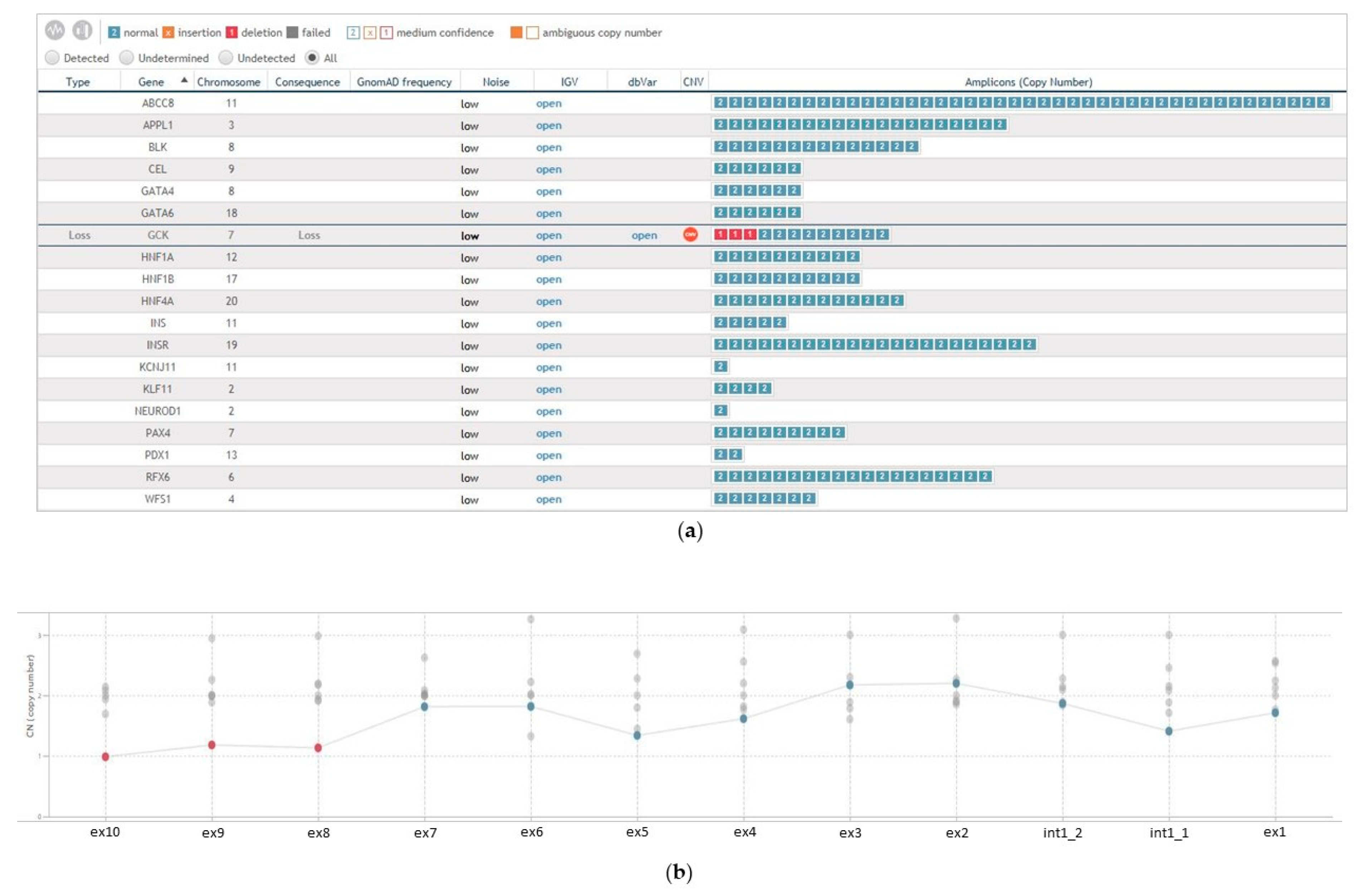
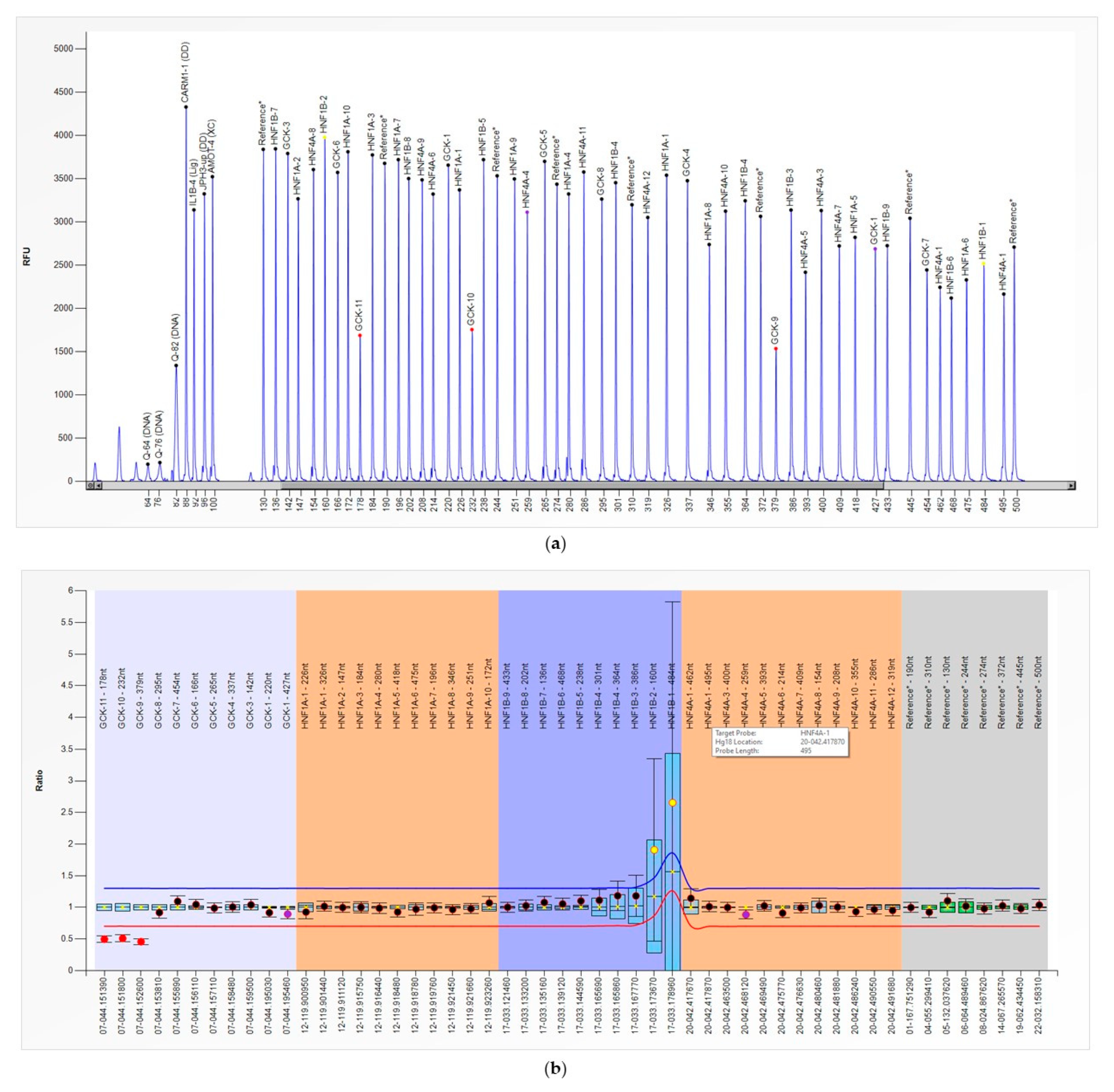
3. Results
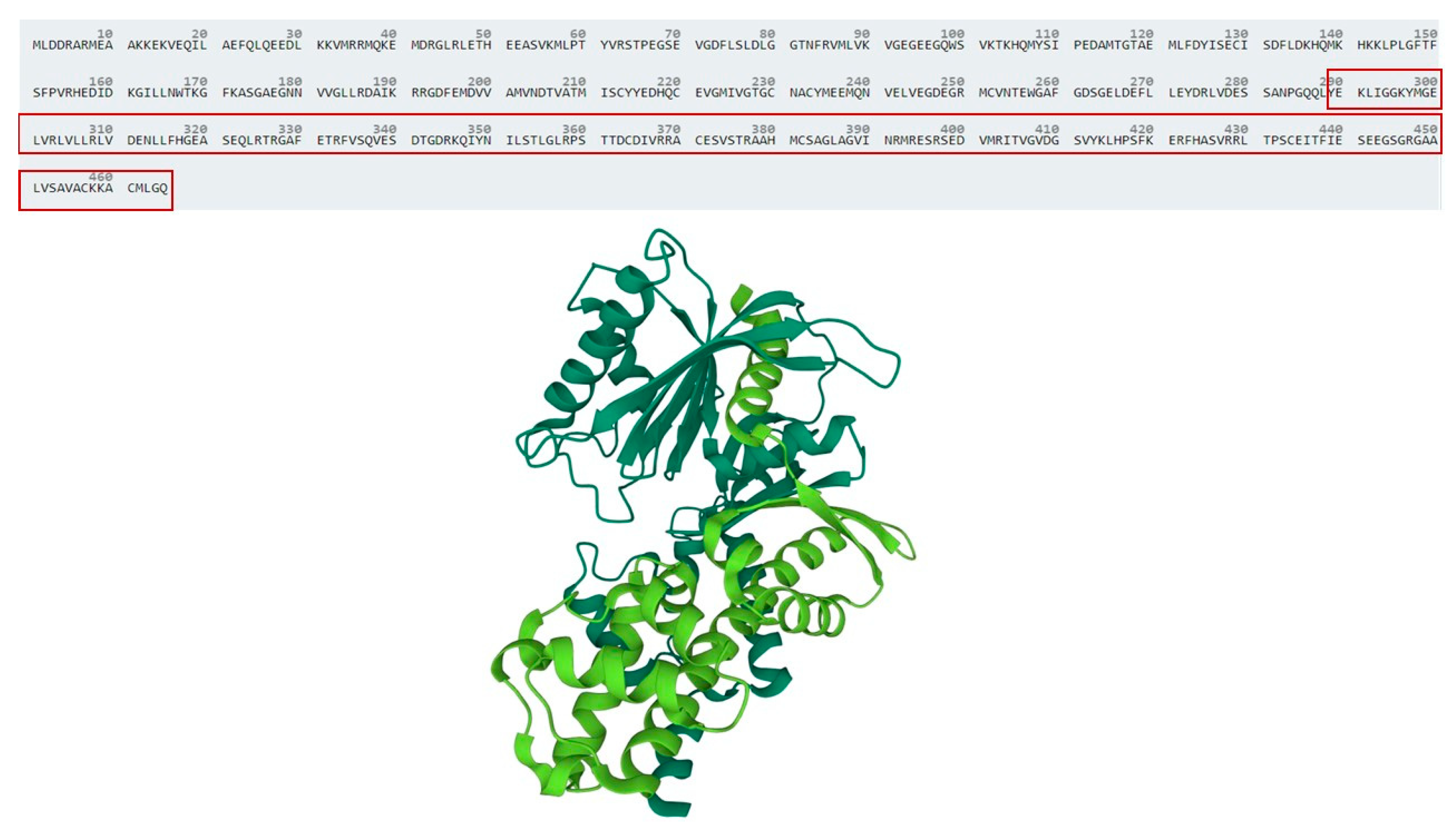
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kavvoura, F.K.; Owen, K.R. Maturity onset diabetes of the young: Clinical characteristics, diagnosis and management. Pediatr. Endocrinol. Rev. 2012, 10, 234–242. [Google Scholar] [PubMed]
- Shepherd, M.; Shields, B.; Hammersley, S.; Hudson, M.; McDonald, T.J.; Colclough, K.; Oram, R.A.; Knight, B.; Hyde, C.; Cox, J.; et al. Systematic Population Screening, Using Biomarkers and Genetic Testing, Identifies 2.5% of the U.K. Pediatric Diabetes Population With Monogenic Diabetes. Diabetes Care 2016, 39, 1879–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nkonge, K.M.; Nkonge, D.K.; Nkonge, T.N. The epidemiology, molecular pathogenesis, diagnosis, and treatment of maturity-onset diabetes of the young (MODY). Clin. Diabetes Endocrino. 2020, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Cabezas, O.; Hattersley, A.T.; Njølstad, P.R.; Mlynarski, W.; Ellard, S.; White, N.; Chi, D.V.; Craig, M.E. International Society for Pediatric and Adolescent Diabetes. ISPAD Clinical Practice Consensus Guidelines 2014. The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr. Diabetes. 2014, 15, 47–64. [Google Scholar] [CrossRef]
- Antal, Z. Maturity-Onset Diabetes of the Young (MODY): Genetic Causes, Clinical Characteristics, Considerations for Testing, and Treatment Options. Endocrines 2021, 2, 485–501. [Google Scholar] [CrossRef]
- Dallali, H.; Pezzilli, S.; Hechmi, M.; Sallem, O.K.; Elouej, S.; Jmel, H.; Ben Halima, Y.; Chargui, M.; Gharbi, M.; Mercuri, L.; et al. Genetic characterization of suspected MODY patients in Tunisia by targeted next-generation sequencing. Acta Diabetol. 2019, 56, 515–523. [Google Scholar] [CrossRef] [Green Version]
- Sperling, M.A.; Garg, A. Monogenic Forms of Diabetes. In Diabetes in America, 3rd ed.; Cowie, C.C., Casagrande, S.S., Menke, A., Cissell, M.A., Eberhardt, M.S., Meigs, J.B., Gregg, E.W., Knowler, W.C., Barrett-Connor, E., Becker, D.J., Eds.; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2018; pp. 1–27. [Google Scholar]
- Jetton, T.L.; Liang, Y.; Pettepher, C.C.; Zimmerman, E.C.; Cox, F.G.; Horvath, K.; Matschinsky, F.M.; Magnuson, M.A. Analysis of upstream glucokinase promoter activity in transgenic mice and identification of glucokinase in rare neuroendocrine cells in the brain and gut. J. Biol. Chem. 1994, 269, 3641–3654. [Google Scholar] [CrossRef]
- Matschinsky, F.M.; Wilson, D.F. The Central Role of Glucokinase in Glucose Homeostasis: A Perspective 50 Years After Demonstrating the Presence of the Enzyme in Islets of Langerhans. Front. Physiol. 2019, 10, 148. [Google Scholar] [CrossRef] [Green Version]
- Christesen, H.B.; Tribble, N.D.; Molven, A.; Siddiqui, J.; Sandal, T.; Brusgaard, K.; Ellard, S.; Njølstad, P.R.; Alm, J.; Jacobsen, B.B.; et al. Activating glucokinase (GCK) mutations as a cause of medically responsive congenital hyperinsulinism: Prevalence in children and characterisation of a novel GCK mutation. Eur. J. Endocrino. 2008, 159, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Esquiaveto-Aun, A.M.; De Mello, M.P.; Paulino, M.F.; Minicucci, W.J.; Guerra-Júnior, G.; De Lemos-Marini, S.H. A new compound heterozygosis for inactivating mutations in the glucokinase gene as cause of permanent neonatal diabetes mellitus (PNDM) in double-first cousins. Diabetol. Metab. Syndr. 2015, 7, 101. [Google Scholar] [CrossRef]
- Froguel, P.; Vaxillaire, M.; Sun, F.; Velho, G.; Zouali, H.; Butel, M.O.; Lesage, S.; Vionnet, N.; Clément, K.; Fougerousse, F.; et al. Close linkage of glucokinase locus on chromosome 7p to early-onset non-insulin-dependent diabetes mellitus. Nature 1992, 356, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Delvecchio, M.; Pastore, C.; Giordano, P. Treatment Options for MODY Patients: A Systematic Review of Literature. Diabetes Ther. 2020, 11, 1667–1685. [Google Scholar] [CrossRef] [PubMed]
- Kant, R.; Davis, A.; Verma, V. Maturity-Onset Diabetes of the Young: Rapid Evidence Review. Am. Fam. Physician 2022, 105, 162–167. [Google Scholar]
- Carmody, D.; Naylor, R.N.; Bell, C.D.; Berry, S.; Montgomery, J.T.; Tadie, E.C.; Hwang, J.L.; Greeley, S.A.W.; Philipson, L.H. GCK-MODY in the US National Monogenic Diabetes Registry: Frequently misdiagnosed and unnecessarily treated. Acta Diabetol. 2016, 53, 703–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina-Ramírez, L.P.; Kyle, C.; Ellingford, J.M.; Wright, R.; Taylor, A.; Bhaskar, S.S.; Campbell, C.; Jackson, H.; Fairclough, A.; Rousseau, A.; et al. Personalised virtual gene panels reduce interpretation workload and maintain diagnostic rates of proband-only clinical exome sequencing for rare disorders. J. Med. Genet. 2022, 59, 393–398. [Google Scholar] [CrossRef]
- Kamata, K.; Mitsuya, M.; Nishimura, T.; Eiki, J.; Nagata, Y. Structural basis for allosteric regulation of the monomeric allosteric enzyme human glucokinase. Structure 2004, 12, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Molnes, J.; Bjørkhaug, L.; Søvik, O.; Njølstad, P.R.; Flatmark, T. Catalytic activation of human glucokinase by substrate binding: Residue contacts involved in the binding of D-glucose to the super-open form and conformational transitions. FEBS J. 2008, 275, 2467–2481. [Google Scholar] [CrossRef] [Green Version]
- Hattersley, A.T.; Turner, R.C.; Permutt, M.A.; Patel, P.; Tanazawa, Y.; Chin, K.C.; O’Rahilly, S.; Watkins, P.J.; Wainscoat, J.S. Linkage of type 2 diabetes to the glucokinase gene. Lancet 1992, 339, 1307–1310. [Google Scholar] [CrossRef]
- Osbak, K.K.; Colclough, K.; Saint-Martin, C.; Beer, N.L.; Bellanné-Chantelot, C.; Ellard, S.; Gloyn, A.L. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum. Mutat. 2009, 30, 1512–1526. [Google Scholar] [CrossRef]
- Ellard, S.; Thomas, K.; Edghill, E.L.; Owens, M.; Ambye, L.; Cropper, J.; Little, J.; Strachan, M.; Stride, A.; Ersoy, B.; et al. Partial and whole gene deletion mutations of the GCK and HNF1A genes in maturity-onset diabetes of the young. Diabetologia 2007, 50, 2313–2317. [Google Scholar] [CrossRef] [Green Version]
- Garin, I.; Rica, I.; Estalella, I.; Oyarzabal, M.; Rodríguez-Rigual, M.; San Pedro, J.I.; Pérez-Nanclares, G.; Fernández-Rebollo, E.; Busturia, M.A.; Castaño, L.; et al. Haploinsufficiency at GCK gene is not a frequent event in MODY2 patients. Clin. Endocrinol. (Oxf.) 2008, 68, 873–878. [Google Scholar] [CrossRef]
- Berberich, A.J.; Huot, C.; Cao, H.; McIntyre, A.D.; Robinson, J.F.; Wang, J.; Hegele, R.A. Copy Number Variation in GCK in Patients With Maturity-Onset Diabetes of the Young. J. Clin. Endocrinol. Metab. 2019, 104, 3428–3436. [Google Scholar] [CrossRef] [PubMed]
- Naik, S.; Riordan-Eva, E.; Thomas, N.S.; Poole, R.; Ashton, M.; Crolla, J.A.; Temple, I.K. Large de novo deletion of 7p15.1 to 7p12.1 involving the imprinted gene GRB10 associated with a complex phenotype including features of Beckwith Wiedemann syndrome. Eur. J. Med. Genet. 2011, 54, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Birkebæk, N.H.; Sørensen, J.S.; Vikre-Jørgensen, J.; Jensen, P.K.; Pedersen, O.; Hansen, T. A De Novo Whole GCK Gene Deletion Not Detected by Gene Sequencing, in a Boy with Phenotypic GCK Insufficiency. Case Rep. Genet. 2011, 2011, 768610. [Google Scholar] [PubMed] [Green Version]
- Yorifuji, T.; Fujimaru, R.; Hosokawa, Y.; Tamagawa, N.; Shiozaki, M.; Aizu, K.; Jinno, K.; Maruo, Y.; Nagasaka, H.; Tajima, T.; et al. Comprehensive molecular analysis of Japanese patients with pediatric-onset MODY-type diabetes mellitus. Pediatr. Diabetes 2012, 13, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Rehm, H.L. Disease-targeted sequencing: A cornerstone in the clinic. Nat. Rev. Genet. 2013, 14, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Niazi, R.; Gonzalez, M.A.; Balciuniene, J.; Evans, P.; Sarmady, M.; Abou Tayoun, A.N. The Development and Validation of Clinical Exome-Based Panels Using ExomeSlicer: Considerations and Proof of Concept Using an Epilepsy Panel. J. Mol. Diagn. 2018, 20, 643–652. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Concolino, P.; Tartaglione, L.; De Paolis, E.; Carrozza, C.; Urbani, A.; Minucci, A.; Pitocco, D.; Santonocito, C. A Novel GCK Large Genomic Rearrangement in a Patient with MODY-2 Detected by Clinical Exome Sequencing. Genes 2022, 13, 2104. https://doi.org/10.3390/genes13112104
Concolino P, Tartaglione L, De Paolis E, Carrozza C, Urbani A, Minucci A, Pitocco D, Santonocito C. A Novel GCK Large Genomic Rearrangement in a Patient with MODY-2 Detected by Clinical Exome Sequencing. Genes. 2022; 13(11):2104. https://doi.org/10.3390/genes13112104
Chicago/Turabian StyleConcolino, Paola, Linda Tartaglione, Elisa De Paolis, Cinzia Carrozza, Andrea Urbani, Angelo Minucci, Dario Pitocco, and Concetta Santonocito. 2022. "A Novel GCK Large Genomic Rearrangement in a Patient with MODY-2 Detected by Clinical Exome Sequencing" Genes 13, no. 11: 2104. https://doi.org/10.3390/genes13112104