

Population Diversity Analysis Provide Insights into Provenance Identification of Dendrobium catenatum
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. RAD Library Development and Sequencing
2.3. SNP Calling
2.4. Phylogenetic Tree Construction
2.5. Population Structure Analyses
2.6. Genetic Diversity and Differentiation
3. Results
3.1. Sequence Data Quality
3.2. Genetic Diversity
3.3. Phylogenetic Tree
3.4. Population Structure
3.5. Genetic Relationships
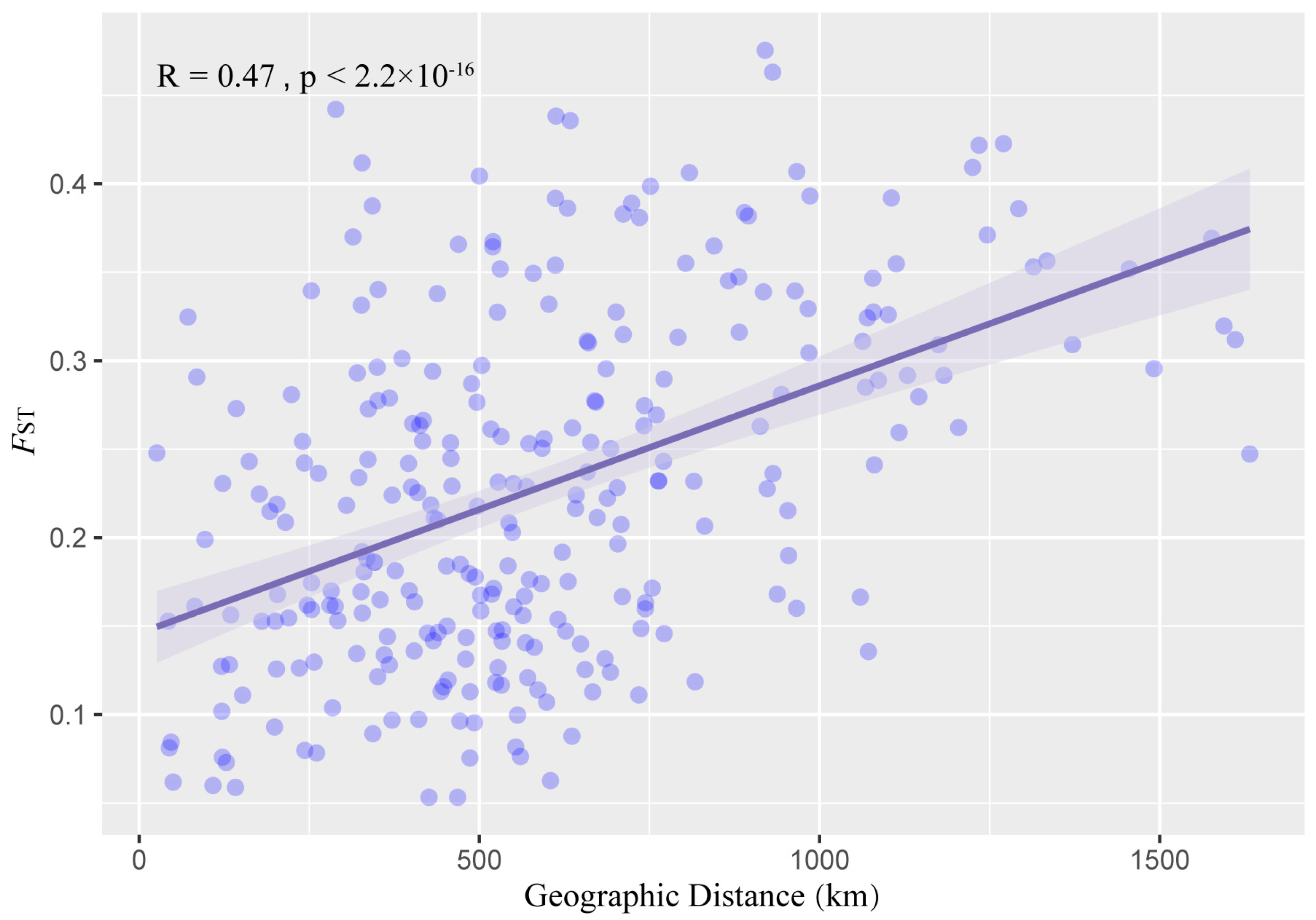
3.6. Genetic Differentiation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cribb, P.; Govaerts, R. Just how many orchids are there? In Proceedings of the 18th World Orchid Conference, Dijon, France, 11–20 March 2005; Rayanl-Roques, A., Roguenant, A., Prat, D., Eds.; Naturalia: Dijon, France, 2005; pp. 161–172. [Google Scholar]
- Pridgeon, A.M.; Cribb, P.J.; Chase, M.W.; Rasmussen, F.N. Genera Orchidacearum Volume 6: Epidendroideae (Part 3); OUP: Oxford, UK, 2014; ISBN 978-0-19-100389-9. [Google Scholar]
- Chinese Pharmacopoeia Commission. Pharmacopoeia of the People’s Republic of China; Chinese Medicine Science and technology Publishing House: Beijing, China, 2020.
- Bao, X.S.; Shun, Q.S.; Chen, L.Z. The Medicinal Plants of Dendrobium (Shi-Hu) in China; Shanghai Medical University Press, Fudan University Press: Shanghai, China, 2001; pp. 56–60. [Google Scholar]
- He, T.G.; Yang, L.T.; Li, Y.R.; Wang, C.Q.; Huang, D.P. Physicochemical properties and antitumor activity of polysaccharide dcpp1a-1 from suspension-cultured protocorms of Dendrobium candidum. Nat. Prod. Res. Dev. 2007, 578–583. [Google Scholar] [CrossRef]
- Liu, X.-F.; Zhu, J.; Ge, S.-Y.; Xia, L.-J.; Yang, H.-Y.; Qian, Y.-T.; Ren, F.-Z. Orally Administered Dendrobium Officinale and Its Polysaccharides Enhance Immune Functions in BALB/c Mice. Nat. Prod. Commun. 2011, 6, 867–870. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Li, Y.; Tao, S.; Wei, G.; Huang, Y.; Chen, D.; Wu, C. Purification, Characterization and Biological Activity of Polysaccharides from Dendrobium Officinale. Molecules 2016, 21, 701. [Google Scholar] [CrossRef] [Green Version]
- Zha, X.Q.; Luo, J.P.; Luo, S.Z.; Jiang, S.T. Structure Identification of a New Immunostimulating Polysaccharide from the Stems of Dendrobium Huoshanense. Carbohydr. Polym. 2007, 69, 86–93. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, Y.; Si, J.-P.; Sun, X.-B.; Sun, G.-B.; Liu, J.-J. A Polysaccharide of Dendrobium Officinale Ameliorates H2O2-Induced Apoptosis in H9c2 Cardiomyocytes via PI3K/AKT and MAPK Pathways. Int. J. Biol. Macromol. 2017, 104, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.-C.; Zha, X.-Q.; Luo, J.-P. A Polysaccharide from Dendrobium Huoshanense Prevents Hepatic Inflammatory Response Caused by Carbon Tetrachloride. Biotechnol. Biotechnol. Equip. 2015, 29, 132–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Q.; Ko, C.-H.; Siu, W.-S.; Li, K.-K.; Wong, C.-W.; Han, X.-Q.; Yang, L.; Lau, C.B.-S.; Hu, J.-M.; Leung, P.-C. Inhibitory Effect of Different Dendrobium Species on LPS-Induced Inflammation in Macrophages via Suppression of MAPK Pathways. Chin. J. Nat. Med. 2018, 16, 481–489. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, J.; Luo, M.; Xie, G.-Y.; Zeng, W.; Wu, Y.; Zhu, Y.; Yang, X.; Guo, A.-Y. Systematic Transcriptome and Regulatory Network Analyses Reveal the Hypoglycemic Mechanism of Dendrobium Fimbriatum. Mol. Ther.-Nucleic Acids 2020, 19, 1–14. [Google Scholar] [CrossRef]
- Pan, L.-H.; Li, X.-F.; Wang, M.-N.; Zha, X.-Q.; Yang, X.-F.; Liu, Z.-J.; Luo, Y.-B.; Luo, J.-P. Comparison of Hypoglycemic and Antioxidative Effects of Polysaccharides from Four Different Dendrobium Species. Int. J. Biol. Macromol. 2014, 64, 420–427. [Google Scholar] [CrossRef]
- Si, J.-P.; Wang, Q.; Liu, Z.-J.; Liu, J.-J.; Luo, Y.-B. Breakthrough in key science and technologies in Dendrobium catenatum industry. China J. Chin. Mater. 2017, 42, 2223–2227. [Google Scholar] [CrossRef]
- Lam, Y.; Ng, T.B.; Yao, R.M.; Shi, J.; Xu, K.; Sze, S.C.W.; Zhang, K.Y. Evaluation of Chemical Constituents and Important Mechanism of Pharmacological Biology in Dendrobium Plants. Evid.-Based Complement. Altern. Med. 2015, 2015, e841752. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.; Guo, Y.; Xu, G.; Xu, L.; An, D. Microscopic ldentification Studies on Dendrobium Stems By Clustering Analysis. J. China Pharm. Univ. 1996, 27, 208–210. [Google Scholar]
- Zha, X.-Q.; Pan, L.-H.; Luo, J.-P.; Wang, J.-H.; Wei, P.; Bansal, V. Enzymatic Fingerprints of Polysaccharides of Dendrobium Officinale and Their Application in Identification of Dendrobium Species. J. Nat. Med. 2012, 66, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Shun, Q.; Huang, Y.; Yang, M.; Chen, L.; Li, M.; Ye, J.; Wang, H.; Jiang, R.; He, Z. Comparative study of hplc characteristic spectrum of Dendrobium officinale from three provenance sources. Tradit. Chin. Drug Res. Clin. Pharmacol. 2014, 25, 467–471. [Google Scholar]
- Huang, L.; Zhang, X.; Chen, S. Research Progress in Quality Ecology of Genuine Regional Drugs. Mod. Tradit. Chin. Med. Mater. Med.-World Sci. Technol. 2019, 21, 844–853. [Google Scholar] [CrossRef]
- Xia, B.; Zhou, Y.; Tan, H.S.; Ding, L.S.; Xu, H.X. Advanced Ultra-Performance Liquid Chromatography–Photodiode Array–Quadrupole Time-of-Flight Mass Spectrometric Methods for Simultaneous Screening and Quantification of Triterpenoids in Poria Cocos. Food Chem. 2014, 152, 237–244. [Google Scholar] [CrossRef]
- Tan, H.-S.; Hu, D.-D.; Song, J.-Z.; Xu, Y.; Cai, S.-F.; Chen, Q.-L.; Meng, Q.-W.; Li, S.-L.; Chen, S.-L.; Mao, Q.; et al. Distinguishing Radix Angelica Sinensis from Different Regions by HS-SFME/GC–MS. Food Chem. 2015, 186, 200–206. [Google Scholar] [CrossRef]
- Zhang, X.; Gu, C.; Ahmad, B.; Huang, L. Optimization of Extract Method for Cynomorium Songaricum Rupr. by Response Surface Methodology. J. Anal. Methods Chem. 2017, 2017, 6153802. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Wang, Z.; Xu, H.; Xu, L.; Zhou, K. Database establishment of the whole rDNA ITS region of Dendrobium species of “fengdou” and authentication by analysis of their sequences. Acta Pharm. Sin. B. 2002, 37, 567–573. [Google Scholar]
- Asahina, H.; Shinozaki, J.; Masuda, K.; Morimitsu, Y.; Satake, M. Identification of Medicinal Dendrobium Species by Phylogenetic Analyses Using matK and rbcL Sequences. J. Nat. Med. 2010, 64, 133–138. [Google Scholar] [CrossRef]
- Liu, H.; Fang, C.; Zhang, T.; Guo, L.; Ye, Q. Molecular Authentication and Differentiation of Dendrobium Species by rDNA ITS Region Sequence Analysis. AMB Expr. 2019, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Ding, X.; Liu, D.; Ding, G.; He, J.; Li, X.; Tang, F.; Chu, B. Intersimple Sequence Repeats (ISSR) Molecular Fingerprinting Markers for Authenticating Populations of Dendrobium Officinale Kimura et Migo. Biol. Pharm. Bull. 2006, 29, 420–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, G.; Xu, G.; Zhang, W.; Lu, S.; Li, X.; Gu, S.; Ding, X.-Y. Preliminary Geoherbalism Study of Dendrobium Officinale Food by DNA Molecular Markers. Eur. Food Res. Technol. 2008, 227, 1283–1286. [Google Scholar] [CrossRef]
- Hodel, R.G.J.; Chen, S.; Payton, A.C.; McDaniel, S.F.; Soltis, P.; Soltis, D.E. Adding Loci Improves Phylogeographic Resolution in Red Mangroves despite Increased Missing Data: Comparing Microsatellites and RAD-Seq and Investigating Loci Filtering. Sci. Rep. 2017, 7, 17598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hale, M.L.; Burg, T.M.; Steeves, T.E. Sampling for Microsatellite-Based Population Genetic Studies: 25 to 30 Individuals per Population Is Enough to Accurately Estimate Allele Frequencies. PLoS ONE 2012, 7, e45170. [Google Scholar] [CrossRef]
- Zimmerman, S.J.; Aldridge, C.L.; Oyler-McCance, S.J. An Empirical Comparison of Population Genetic Analyses Using Microsatellite and SNP Data for a Species of Conservation Concern. BMC Genom. 2020, 21, 382. [Google Scholar] [CrossRef]
- Fischer, M.C.; Rellstab, C.; Leuzinger, M.; Roumet, M.; Gugerli, F.; Shimizu, K.K.; Holderegger, R.; Widmer, A. Estimating Genomic Diversity and Population Differentiation—An Empirical Comparison of Microsatellite and SNP Variation in Arabidopsis Halleri. BMC Genom. 2017, 18, 69. [Google Scholar] [CrossRef] [Green Version]
- Shendure, J.; Ji, H. Next-Generation DNA Sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef]
- Ganal, M.W.; Altmann, T.; Roder, M.S. SNP Identification in Crop Plants. Curr. Opin. Plant Biol. 2009, 12, 211–217. [Google Scholar] [CrossRef]
- Ma, B.; Liao, L.; Peng, Q.; Fang, T.; Zhou, H.; Korban, S.S.; Han, Y. Reduced Representation Genome Sequencing Reveals Patterns of Genetic Diversity and Selection in Apple. J. Integr. Plant Biol. 2017, 59, 190–204. [Google Scholar] [CrossRef]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-Wide Genetic Marker Discovery and Genotyping Using next-Generation Sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Andolfatto, P.; Davison, D.; Erezyilmaz, D.; Hu, T.T.; Mast, J.; Sunayama-Morita, T.; Stern, D.L. Multiplexed Shotgun Genotyping for Rapid and Efficient Genetic Mapping. Genome Res. 2011, 21, 610–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Li, M.; Hu, X.; Ma, Q.; Mu, Y.; Tan, Z.; Xia, Q.; Zhang, G.; Nian, H. Construction of High-Density Genetic Map and QTL Mapping of Yield-Related and Two Quality Traits in Soybean RILs Population by RAD-Sequencing. BMC Genom. 2017, 18, 466. [Google Scholar] [CrossRef] [PubMed]
- Valdisser, P.A.M.R.; Pappas, G.J.; de Menezes, I.P.P.; Müller, B.S.F.; Pereira, W.J.; Narciso, M.G.; Brondani, C.; Souza, T.L.P.O.; Borba, T.C.O.; Vianello, R.P. SNP Discovery in Common Bean by Restriction-Associated DNA (RAD) Sequencing for Genetic Diversity and Population Structure Analysis. Mol. Genet. Genom. 2016, 291, 1277–1291. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yin, S.; Wu, L.; Dai, D.; Wang, H.; Liu, C.; Tang, L. Genetic Diversity and Structure of Wild and Cultivated Amorphophallus Paeoniifolius Populations in Southwestern China as Revealed by RAD-Seq. Sci. Rep. 2017, 7, 14183. [Google Scholar] [CrossRef] [Green Version]
- Hirase, S.; Tezuka, A.; Nagano, A.J.; Kikuchi, K.; Iwasaki, W. Genetic Isolation by Distance in the Yellowfin Goby Populations Revealed by RAD Sequencing. Ichthyol. Res. 2020, 67, 98–104. [Google Scholar] [CrossRef]
- Martin Cerezo, M.L.; Kucka, M.; Zub, K.; Chan, Y.F.; Bryk, J. Population Structure of Apodemus Flavicollis and Comparison to Apodemus Sylvaticus in Northern Poland Based on RAD-Seq. BMC Genom. 2020, 21, 241. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.-Q.; Xu, Q.; Bian, C.; Tsai, W.-C.; Yeh, C.-M.; Liu, K.-W.; Yoshida, K.; Zhang, L.-S.; Chang, S.-B.; Chen, F.; et al. The Dendrobium Catenatum Lindl. Genome Sequence Provides Insights into Polysaccharide Synthase, Floral Development and Adaptive Evolution. Sci. Rep. 2016, 6, 19029. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Cornelis, A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast Model-Based Estimation of Ancestry in Unrelated Individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Catchen, J.; Hohenlohe, P.A.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An Analysis Tool Set for Population Genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, B.; Ulrich Wittelsbürger; Ramos-Onsins, S.E.; Lercher, M.J. PopGenome: An Efficient Swiss Army Knife for Population Genomic Analyses in R. Mol. Biol. Evol. 2014, 31, 1929–1936. [Google Scholar] [CrossRef] [Green Version]
- Chi, Z.; Shan-Shan, D.; Jun-Yang, X.; Wei-Ming, H.; Tie-Lin, Y. PopLDdecay: A Fast and Effective Tool for Linkage Disequilibrium Decay Analysis Based on Variant Call Format Files. Bioinformatics 2018, 35, 1786–1788. [Google Scholar] [CrossRef]
- Bay, R.A.; Harrigan, R.J.; Underwood, V.L.; Gibbs, H.L.; Smith, T.B.; Ruegg, K. Genomic Signals of Selection Predict Climate-Driven Population Declines in a Migratory Bird. Science 2018, 359, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Hou, J.; Fu, N.; Wei, M.; Li, Y.; Yu, K.; Song, H.; Li, S.; Liu, J. Identification of QTL Related to Anther Color and Hull Color by RAD Sequencing in a RIL Population of Setaria Italica. BMC Genom. 2021, 22, 556. [Google Scholar] [CrossRef]
- Sunde, J.; Yıldırım, Y.; Tibblin, P.; Forsman, A. Comparing the Performance of Microsatellites and RADseq in Population Genetic Studies: Analysis of Data for Pike (Esox Lucius) and a Synthesis of Previous Studies. Front. Genet. 2020, 11, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohling, J.; Small, M.; Von Bargen, J.; Louden, A.; DeHaan, P. Comparing Inferences Derived from Microsatellite and RADseq Datasets: A Case Study Involving Threatened Bull Trout. Conserv. Genet. 2019, 20, 329–342. [Google Scholar] [CrossRef]
- Li, X.; Ding, X.; Chu, B.; Zhou, Q.; Ding, G.; Gu, S. Genetic Diversity Analysis and Conservation of the Endangered Chinese Endemic Herb Dendrobium Officinale Kimura et Migo (Orchidaceae) Based on AFLP. Genetica 2008, 133, 159–166. [Google Scholar] [CrossRef]
- Ding, G.; Ding, X.; Shen, J.; Tang, F.; Liu, D.; He, J.; Li, X.; Chu, B. Genetic diversity and molecular authentication of wild populations of Dendrobium officinale by RAPD. Yao Xue Xue Bao 2005, 40, 1028–1032. [Google Scholar] [PubMed]
- Gupta, S.K.; Baek, J.; Carrasquilla-Garcia, N.; Penmetsa, R.V. Genome-Wide Polymorphism Detection in Peanut Using next-Generation Restriction-Site-Associated DNA (RAD) Sequencing. Mol. Breed. 2015, 35, 145. [Google Scholar] [CrossRef]
- Jiao, Y.; Zhao, H.; Ren, L.; Song, W.; Zeng, B.; Guo, J.; Wang, B.; Liu, Z.; Chen, J.; Li, W.; et al. Corrigendum: Genome-Wide Genetic Changes during Modern Breeding of Maize. Nat. Genet. 2014, 46, 1039–1040. [Google Scholar] [CrossRef] [Green Version]
- Dillon, M.M.; Sung, W.; Lynch, M.; Cooper, V.S. The Rate and Molecular Spectrum of Spontaneous Mutations in the GC-Rich Multichromosome Genome of Burkholderia Cenocepacia. Genetics 2015, 200, 935–946. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Liu, L.; Wang, Y.; Zhang, Q.; Fan, G.; Zhang, S.; Wang, Y.; Liao, K. Genetic Diversity, Population Structakure, and Relationships of Apricot (Prunus) Based on Restriction Site-Associated DNA Sequencing. Hortic. Res. 2020, 7, 69. [Google Scholar] [CrossRef]
- Ye, M.; Wang, X.; Zhou, Y.; Huang, S.; Liu, A. Genetic Diversity and Population Structure of Cultivated Dendrobium Huoshanense (C.Z. Tang et S.J. Cheng) Using SNP Markers Generated from GBS Analysis. Pak. J. Bot 2021, 53, 1683–1690. [Google Scholar] [CrossRef]
- Hou, B.; Luo, J.; Zhang, Y.; Niu, Z.; Xue, Q.; Ding, X. Iteration Expansion and Regional Evolution: Phylogeography of Dendrobium Officinale and Four Related Taxa in Southern China. Sci. Rep. 2017, 7, 43525. [Google Scholar] [CrossRef] [Green Version]
- Hinsley, A.; de Boer, H.J.; Fay, M.F.; Gale, S.W.; Gardiner, L.M.; Gunasekara, R.S.; Kumar, P.; Masters, S.; Metusala, D.; Roberts, D.L.; et al. A Review of the Trade in Orchids and Its Implications for Conservation. Bot. J. Linn. Soc. 2018, 186, 435–455. [Google Scholar] [CrossRef]
- Niu, Z.; Hou, Z.; Wang, M.; Ye, M.; Zhang, B.; Xue, Q.; Liu, W.; Ding, X. A Comparative Plastomics Approach Reveals Available Molecular Markers for the Phylogeographic Study of Dendrobium Huoshanense, an Endangered Orchid with Extremely Small Populations. Ecol. Evol. 2020, 10, 5332–5342. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Location | Specimen Code | Population Code | Individual Number |
|---|---|---|---|---|
| D. catenatum | Guangze, Fujian Province | Z.J. Liu 10572 | FJGZTP01 | 5 |
| Liancheng, Fujian Province | Z.J. Liu 10583 | FJLCTP02 | 4 | |
| Pingjiang, Hunan Province | Z.J. Liu 10580 | HNPJTP04 | 5 | |
| Yiyang, Hunan Province | Z.J. Liu 10582 | HNYYTP05 | 5 | |
| Yuanling, Hunan Province | Z.J. Liu 10578 | HNYLTP06 | 5 | |
| Xinning, Hunan Province | Z.J. Liu 9506 | HNXNTP36 | 3 | |
| Lipu, Guangxi Province | Z.J. Liu 10574 | GXLPTP07 | 5 | |
| Gongcheng, Guangxi Province | Z.J. Liu 9497 | GXGCTP09 | 4 | |
| Xing’an, Guangxi Province | Z.J. Liu 9518 | GXXATP10 | 5 | |
| Lishui, Zhengjiang Province | Z.J. Liu 9520 | ZJLSTP12 | 5 | |
| Wuyi, Zhengjiang Province | Z.J. Liu 9511 | ZJWYTP32 | 5 | |
| Quzhou, Zhengjiang Province | Z.J. Liu 9510 | ZJQZTP13 | 5 | |
| Yanshan, Jiangxi Province | Z.J. Liu 10576 | JXQSTP14 | 5 | |
| Xiushui, Jiangxi Province | Z.J. Liu 10573 | JXXSTP15 | 4 | |
| Huichang, Jiangxi Province | Z.J. Liu 10581 | JXHCTP16 | 5 | |
| Xingguo, Jiangxi Province | Z.J. Liu 9517 | JXXGTP17 | 4 | |
| Nanxiong, Guangdong Province | Z.J. Liu 10584 | GDNXTP19 | 5 | |
| Heyuan, Guangdong Province | Z.J. Liu 10585 | GDHYTP20 | 4 | |
| Shaoguan, Guangdong Province | Z.J. Liu 9504 | GDSGTP22 | 5 | |
| Qujing, Yunnan Province | Z.J. Liu 10579 | YNQJTP24 | 4 | |
| Wangzishan, Yunnan Province | Z.J. Liu 7462 | WZSTP34 | 4 | |
| Xianning, Hubei Province | Z.J. Liu 9514 | HBXNTP25 | 5 | |
| Sandu, Guizhou Province | Z.J. Liu 11147 | GZSDTP29 | 4 | |
| Dushan, Guizhou Province | Z.J. Liu 11148 | GZDSTP30 | 4 | |
| D. huoshanense | Huangshan, Anhui Province | Z.J. Liu 9508 | AHHSHS26 | 5 |
| Longhushan, Jiangxi Province | Z.J. Liu 9500 | JXLHSHS28 | 5 |
| Taxon | Population | AP | HO | HE | π | FIS | Tajima’s D |
|---|---|---|---|---|---|---|---|
| D. catenatum | 0.0992 | 0.1575 | 0.1584 | 0.3836 | |||
| FJGZTP01 | 3254 | 0.0786 | 0.0969 | 0.1165 | 0.0725 | 0.6534 | |
| FJLCTP02 | 1934 | 0.0729 | 0.0833 | 0.099 | 0.0496 | 0.3964 | |
| HNPJTP04 | 2871 | 0.0895 | 0.1131 | 0.1297 | 0.0824 | 0.4221 | |
| HNYYTP05 | 4263 | 0.0818 | 0.1082 | 0.1223 | 0.084 | 0.4411 | |
| HNYLTP06 | 4689 | 0.0976 | 0.1196 | 0.1371 | 0.081 | 0.5556 | |
| GXLPTP07 | 5512 | 0.0952 | 0.1312 | 0.1484 | 0.1135 | 0.4684 | |
| GXGCTP09 | 2679 | 0.0965 | 0.1041 | 0.1355 | 0.066 | 0.7907 | |
| GXXATP10 | 2940 | 0.0724 | 0.0891 | 0.1074 | 0.0628 | 0.8556 | |
| ZJLSTP12 | 2355 | 0.0994 | 0.1182 | 0.1341 | 0.0737 | 0.3298 | |
| ZJQZTP13 | 3084 | 0.1123 | 0.1105 | 0.1245 | 0.0256 | 0.3962 | |
| JXQSTP14 | 3844 | 0.123 | 0.1115 | 0.1243 | 0.0053 | 0.3384 | |
| JXXSTP15 | 2552 | 0.0783 | 0.107 | 0.1257 | 0.0922 | 0.3767 | |
| JXHCTP16 | 3417 | 0.0778 | 0.1037 | 0.1188 | 0.0866 | 0.3404 | |
| JXXGTP17 | 4215 | 0.0919 | 0.096 | 0.1134 | 0.039 | 0.5287 | |
| GDNXTP19 | 3515 | 0.0848 | 0.1022 | 0.1154 | 0.0659 | 0.3163 | |
| GDHYTP20 | 5221 | 0.1019 | 0.1155 | 0.133 | 0.0638 | 0.2443 | |
| GDSGTP22 | 3845 | 0.1078 | 0.0862 | 0.0961 | −0.0208 | 0.4748 | |
| YNQJTP24 | 3933 | 0.1144 | 0.1153 | 0.1343 | 0.039 | 0.4327 | |
| HBXNTP25 | 2757 | 0.0979 | 0.1057 | 0.1185 | 0.0426 | 0.5005 | |
| GZSDTP29 | 4461 | 0.0975 | 0.1147 | 0.1326 | 0.0696 | 0.4221 | |
| GZDSTP30 | 9655 | 0.1516 | 0.1121 | 0.1288 | −0.0434 | 0.4646 | |
| ZJWYTP32 | 2932 | 0.0885 | 0.106 | 0.1191 | 0.0664 | 0.2539 | |
| WZSTP34 | 4820 | 0.125 | 0.1165 | 0.1342 | 0.0208 | 0.4762 | |
| HNXNTP36 | 2967 | 0.1034 | 0.0994 | 0.1218 | 0.0332 | 0.4315 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.-Y.; Li, T.-Z.; Zheng, F.; Chen, J.-B.; Yan, Y.-H.; Huang, J.-X. Population Diversity Analysis Provide Insights into Provenance Identification of Dendrobium catenatum. Genes 2022, 13, 2093. https://doi.org/10.3390/genes13112093
Wu X-Y, Li T-Z, Zheng F, Chen J-B, Yan Y-H, Huang J-X. Population Diversity Analysis Provide Insights into Provenance Identification of Dendrobium catenatum. Genes. 2022; 13(11):2093. https://doi.org/10.3390/genes13112093
Chicago/Turabian StyleWu, Xin-Yi, Ting-Zhang Li, Fang Zheng, Jian-Bing Chen, Yue-Hong Yan, and Jiu-Xiang Huang. 2022. "Population Diversity Analysis Provide Insights into Provenance Identification of Dendrobium catenatum" Genes 13, no. 11: 2093. https://doi.org/10.3390/genes13112093